
Complete tutorial in simple terms
How to predict protein structures with AlphaFold 2 using ColabFold
Because many readers reach out asking about this, here’s a guide to AlphaFold including a straightforward introduction and how to run it through ColabFold including ways to model multimers, employ custom templates, and interpret 1D and 2D confidence metrics -all kept simple and with a real-life example.
Article by a former CASP assessor who uses molecular modeling and simulations in its work at EPFL. To see more technical articles, check this index.
Introduction
The structure of a protein, that is the spatial disposition of all its atoms, determines its function, interaction with other molecules, regulation, stability, and all other kinds of relevant properties. Knowing a protein’s structure is therefore essential to understand how it works, how we can block or activate its activity with a drug, how we can tune its function or stability for a biotechnological application, etc. That’s all relevant as fundamental knowledge and for applications in medicine and processes.
Protein structure modeling is the process of predicting the three-dimensional structure of a protein (the “target” protein) from its amino acid sequence. Methods for protein structure prediction are essential in modern biology, because a huge number of proteins are not amenable to the kind of experimentation required to solve their structures: some cannot be produced in stable forms in solution, others can be produced but they they do not crystallize, or they are too small for some techniques, or too big for others, etc.
There are several methods available for protein structure modeling, that can be classified mainly depending on the availability of structures for (presumably) similar proteins that can be used as “templates”. When such similar structures are at hand, so-called “homology modeling” is the method to go. In a procedure for homology modeling, one or more known structures are used as the core “structural information” from which models are built for the target protein. The templates are typically derived from the Protein Data Bank (PDB), a database of experimentally determined protein structures; however, they could well be structures that you’ve solved but haven’t released, or even other models built previously.
Homology modeling has been around for some decades already, and it works reasonably to very well when good templates are available -that is, structures are available for proteins that are quite similar to the target in terms of both sequence and structure.
But what happens when no good templates are at hand?
If no suitable templates are available in the PDB, so-called ab initio modeling can be used. This is not just one method but actually a broad range of computational techniques to predict protein structures from scratch, without the need for templates. Most methods for ab initio modeling are computationally intensive and typically produces less accurate results than homology modeling. Or that’s how it was until AlphaFold 2 (and newer methods that I’ve also discussed, such as ESMFold) showed up.
CASP, Deepmind, and AlphaFold
CASP (Critical Assessment of Techniques for Protein Structure Prediction) is a biennial scientific experiment that evaluates the performance of protein structure prediction methods. The experiment aims to advance the field of protein structure prediction by providing a rigorous and fair evaluation of different methods, as well as to identify areas where further research is needed.
DeepMind, a subsidiary of Alphabet Inc., participated in the CASP13 experiment in 2018 with a protein structure prediction method called AlphaFold. This method won CASP13 by a small margin, not really inventing anything new but engineering the most out of the state of the art. at that time. The success of this program sparked further research into the use of machine learning for protein structure prediction and opened up new possibilities for advancing the field… especially with AlphaFold 2 which, contrary to AlphaFold 1, was a true ground-breaker in the field, changing biology forever.
AlphaFold 2 is as of today the state-of-the-art deep learning system for protein structure prediction. It was unveiled in November 2020 as the first system that can accurately predict protein structures with near-atomic resolution even in the absence of templates. AlphaFold 2's architecture combines various kinds of neural networks trained on a huge representative dataset of the PDB. Given an input sequence for a target protein to be modeled, AlphaFold predicts its 3D structure and two different of quality metrics -all of which we cover here. AlphaFold 2 actually does use templates if available; and besides, it uses alignments of protein sequences similar to the target. Both affect the quality of the results and also how you run the program, as we’ll see later on in this tutorial.
AlphaFold 2 has been shown to outperform existing methods for protein structure prediction and has been used to solve several challenges related to the problem of protein structure prediction itself. Besides, it triggered a revolution in structural biology and structural bioinformatics.
For more on AlphaFold 2 before we delve into ColabFold and the tutorial, see this index to all my articles on CASP and AlphaFold, and in particular this article which covers all kinds of applications for which it has been exploited beyond the core use in protein structure prediction itself:
ColabFold’s implementations of AlphaFold 2 allow you to run it very easily yet with full control
To run a protein of your interest in AlphaFold 2, you would need to somehow get a handle on an executable version of the program, and then provide it with the amino acid sequence of the protein whose structure you want to predict. AlphaFold can use just this information to make a prediction of the protein’s 3D structure, together with model quality metrics.
But with just that, you’d be missing some elements that are essential to boost the chances of getting good models out. Indeed, AlphaFold 2 can “optionally” use templates in the PDB and/or multiple sequence alignments of proteins related to the query sequence you are modeling. But I wrote “optionally” in quotes because in practice these options are very important, in many cases essential, to achieve high-quality predictions. Therefore, they are highly recommended.
How to get a grasp on the AlphaFold program, and how to do efficient sequence and template searches? ColabFold solves both problems for you in the simplest manner. Indeed it solves them so well, that ColabFold represents today the most widely used way of accessing AlphaFold!
ColabFold is a suite of online-running notebooks that seamlessly integrate a running version of AlphaFold 2 (or its version adapted for complexes, “AlphaFold multimer”) and efficient ways to get templates and build MSAs by using a server called MMseqs2, developed by some of the very ColabFold developers.
While the above paragraphs served as a quick introduction to why we’ll use here AlphaFold 2 through ColabFold, you can know more about all this in this blog entry and in a peer-reviewed article published by its authors:
Tutorial to run AlphaFold using ColabFold on Google-provided hardware
0Before you start, you need to set up a Google account in order to access and use these programs. If you have an account on Gmail, YouTube, etc. then you already have a Google account!
1Now connect to the right Google Colab notebook you need depending on how you want to run AlphaFold. By “depending on how you want to run AlphaFold”, I mean the following:
If you go to the official ColabFold page:
You will find a table of supported notebooks. These are the Release versions as of mid-January 2023:

You can see you have 3 flavors of AlphaFold, plus RoseTTAFold and ESMFold. RoseTTAFold was the immediate academic response to AlphaFold 2 right at the same time it came out, and ESMFold is the AI tool for protein structure prediction using deep protein language models, released recently by Meta (that I covered here).
Let’s look briefly at the three AlphaFold notebooks:
- “from Deepmind” was the first-ever tool available to run AlphaFold, released by Deepmind together with AlphaFold’s source code and papers. Although you could perfectly use it to model structures, it is certainly superseded by the more powerful AlphaFold2_mmseqs2.
- “AlphaFold2_mmseqs2” performs protein structure and complex prediction using AlphaFold or Alphafold2-multimer, respectively (AlphaFold-multimer being a tweaked version of AlphaFold that is slightly better than regular AlphaFold 2 at modeling complexes between multiple proteins). In this notebook, sequence alignments and template detection are carried out by the programs MMseqs2 and HHsearch.
- “AlphaFold2_batch” is simply a batch implementation of the above notebook, useful to run predictions on several sequences in a single shot.
For most of your work in Colab Notebooks using Google’s resources, you will likely use AlphaFold2_mmseqs2. In particular, note that although AlphaFold2_batch allows you to run multiple predictions in a single session, Colab Notebooks running on Google’s resources are in practice halted after some time, so this won’t help you model large numbers of proteins in a single run -should suffice for a bunch, though. I found the batch mode actually useful if it is downloaded run locally. Here, I willl cover the use of AlphaFold2_mmseqs2.
2To run AlphaFold2_mmseqs2 in Google Colab using Google-provided hardware consists in setting up the target sequence/s for modeling, and some choices regarding templates and other options.
See this example, where I’m setting up a run for a homodimer, i.e. folding two copies of a protein together into a binary complex (see the colon separating the first from the second sequence). This example is interesting because the actual experimental structure was just released, so AlphaFold doesn’t know about it, hence we can compare its predictions to the “true” structure. Besides, the structure has a quite peculiar topology, as you will see.
In this example I’m asking AlphaFold 2 to use a custom template (see arrow), contrary to just letting it find templates automatically at the PDB (option “PDB70”). Out of the 600+ residues of the target sequence, this template covers the last 300 -important for the analysis stage because we expect at least this part to be modeled correctly.
I also tell the program to save the files to my Google drive (arrow in “Advanced settings”):

3Simply click “Run all” in the “Runtime” menu. You’ll immediately see this screen, on which you have to say “Run anyway”:

If you chose to add your own templates as I did here, you must click a “Browse” button to upload the PDB of your custom template:
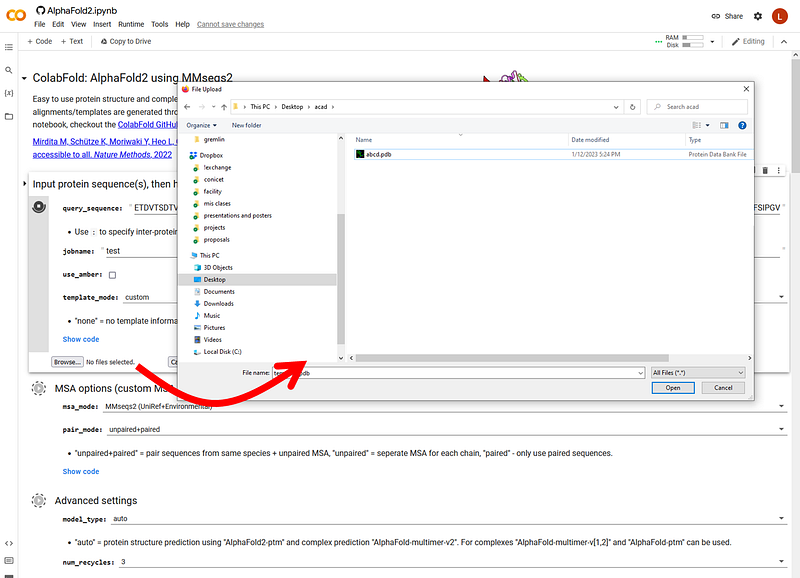
4Now, just let it run. You’ll see the progress as it goes, first installing dependencies (don’t worry, nothing actually goes to your disk, it’s all online!), downloading the weights of the network, getting sequences for the alignment (and grabbing templates from the PDB if you chose the PDB70 option):

Immediately after that, the program starts to craft the first model. After some minutes you’ll see two 2D views of this first model (left, colored by chain; right, colored by pLDDT which is a confidence metric per residue where blue is better) together with some basic confidence metrics:
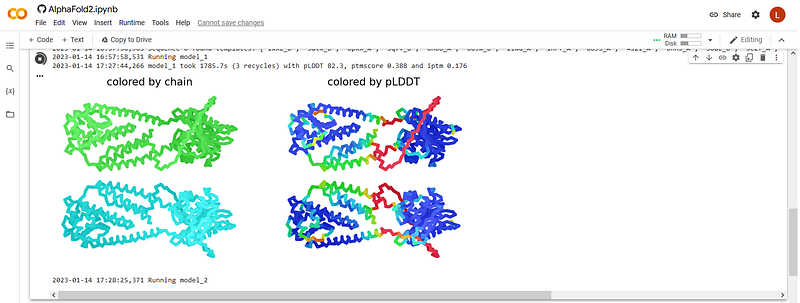
This first output already gives us some hints on how the modeling procedure will go. First, the global quality metrics are good but not very high: pLDDT=82.3, pTMscore=0.388, and ipTM = 0.176; in particular, the latter two are rather low. The modeling so far seems to be saying that the two copies of the protein don’t interact with each other, hence according to one would conclude that this protein is probably not dimeric but monomeric, contrary to our assumption of dimeric character when we set it to be modeled as such (spoiler: we now know that this protein actually is dimeric… AlphaFold seems to not be catching this… the program is not infallible!).
What else can we say from this first model? Well there’s a lot or red, so there’s a big chunk that AlphaFold doesn’t know how to model -or it might actually be disordered, we don’t know. On the other hand, two regions are dominated by cyan and blue, so we can be quite sure these domains are probably modeled correctly -anyway we’ll know more about this at the end of the run. In-between, the green helicesare probably real but the program doesn’t know where to place them exactly; but again, we’ll know more about this at the end of the run.
5 Analyze the full results once the run is over. When AlphaFold has finished running, it will present you a 3D representation that you can view in-place or download as PDB file for inspection in your program of choice; plus a set of graphics with very relevant information.
Here’s the model that AlphaFold ranked top among the five it produced, shown in PyMOL color-coded by its B-factor column which consists in the residue-wise pLDDT score -also present in one of the plots as discussed next:

As anticipated, there are two domains quite well modeled (blue), a long loop that AlphaFold 2 cannot model confidently (or might actually be disordered, red), and some helices of dubious placement (white-ish). That the upper domains are well modeled is expected, because they are very well covered by the templates I provided. That the other domain is quite blue means that AlphaFold probably found structural patterns from its internal knowledge about protein structure, that it could extrapolate to our target.
You can also see the pLDDTs in the plot on the bottom right of this screenshot from ColabFold. There are five traces, one per model and all very similar. On the x axis (residue) the pattern seems duplicated; this is because it corresponds to the two copies of the same protein, that we modeled together in an attempt to predict a homodimer:

The other very important plot is the set of 2D matrices in the top, so-called PAE plots after “Predicted Alignment Error”. The PAE is the RMSD predicted for each possible pair of residues if it were to be aligned to the “true” structure. You want it all to be blue and nothing red. There’s one PAE plot for each of the 5 models produced, just like there are 5 traces in the pLDDT plots.
In this output, all PAE plots present some blue only around the diagonal, being all very red outside of it. This indicates that each domain of each protein is expected to be quite well modeled (mostly blue), but the relative arrangement of the domains is likely off (white and some red). Looking in more detail, you can see that the second domain of each protein is “more blue”; this is to expect because we provided a template for this domain.
The other important piece of information we can get from the PAE plots is that the relative arrangement of the two proteins lacks any support at all, and is likely just plain wrong, as it is all deep red.
The third plot, on the bottom left, indicates how deep the obtained alignments are at each residue of the proteins (that is, how many sequences include each residue). You want this to be as high as possible, ideally covering the whole proteins smoothly. In this case it’s not bad for the C-terminal domains, but it is a bit poor for the N-terminal ones.
Comparing the results to the actual structure!
The example I chose is especially interesting because the actual structure is known but it was just released last week, so AlphaFold doesn’t know about it. And it wasn’t even among the PDB-based templates from its last update, had I chosen to use them.
So, we can compare the predictions we got to the actual structures, as in a real-life exercise.
Let’s do this comparison in stages. First, the overall topology: the experimental structure revealed that the protein is in fact dimeric; moreover, it dimerizes by “domain swapping” which means that parts of each molecule fold together with parts of the other. See for example here PDB 8EXP color-coded by chain, one in grey and one in magenta (and compare with the models shown above):

In the next picture you can see this again but with one protein colored in rainbow mode from the N-terminus in blue to the C-terminus in red:
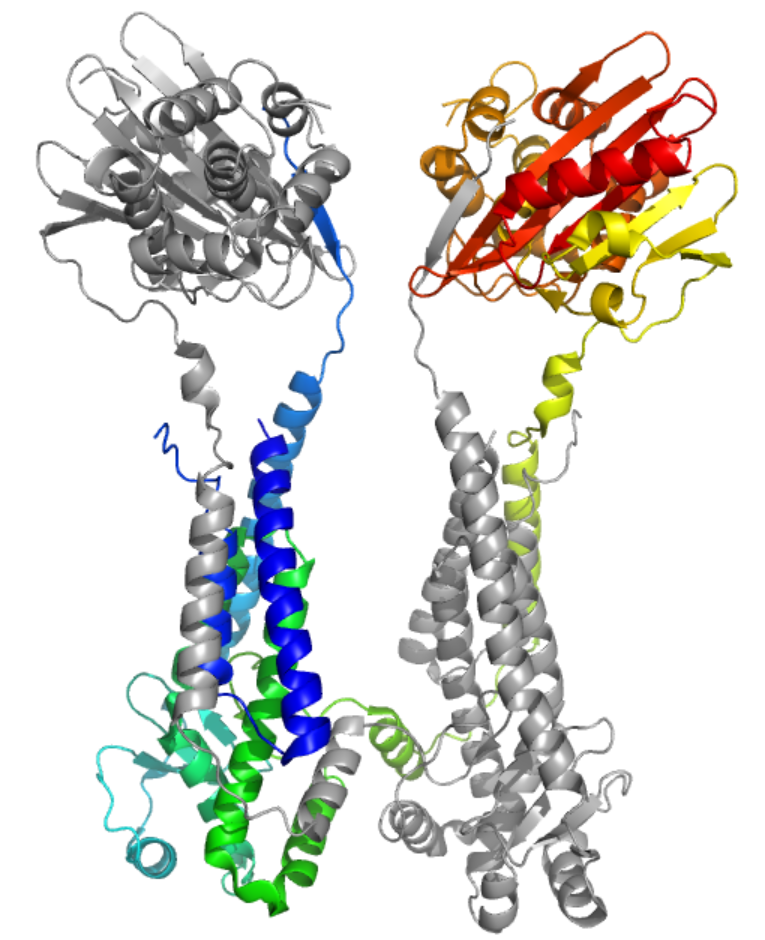
Although this may look like a total failure, there’s quite some merit to how AlphaFold modeled the big chunk for which there are no good templates in the PDB. The next picture focuses specifically on this domain. It shows the model color-coded by pLDDT, all quite blue which means quite high confidence, and the corresponding portion of the experimental structure in grey. The match is quite remarkable, as you can see in the superposition on the left:

Thus, if one wanted to design biochemical experiments to probe how this specific part of the protein works, the model would have been quite helpful!
Conclusion
You’ve seen here how to use AlphaFold in ColabFold in quite some detail, exploring several of its features. One important point is that despite the AlphaFold model is not entirely correct, it would still have helped to guide biology, because the overall topology and even details within each domain are quite good. I mean this even considering that the dimeric nature is not picked up by the model, because despite not all correct, unswapping the swapped elements makes models even closer to the experimental structure.
Hopefully, besides teaching you how to use the program, this real-life example (i.e. modeling a protein without templates, that we can actually evaluate because there is a structure) taught you how to interpret the quality estimate plots and also how to treat the structural models: not as the absolute truth, but probably having at least some useful elements.
Additional resources
This other tutorial is a bit less detailed but is very interesting too:
The main ColabFold paper:
Other articles by me in the same style
www.lucianoabriata.com I write and photoshoot about everything that lies in my broad sphere of interests: nature, science, technology, programming, etc. Become a Medium member to access all its stories (affiliate links of the platform for which I get small revenues without cost to you) and subscribe to get my new stories by email. To consult about small jobs, check my services page here. You can contact me here.




