
Can we predict not only static protein structures but also their structural diversity?
A recent work applying AlphaFold 2 in a special way suggests this could be possible.
Background:
It’s been almost one year since AlphaFold 2’s paper and code were released by Google / Deepmind, and new things keep coming out. I’ve been keeping track about this in various articles here on Medium, including a just-published opinion about what will happen next:
Now, a paper in eLife explored a question prompted by the observation that AlphaFold 2 had predicted multiple valid structures for one of its CASP14 targets. In simple terms, the question was: given that AlphaFold could predict multiple valid structures for this protein, so far anecdotally, can we manipulate it to achieve the same more systematically on other proteins?
The answer is: probably yes.
The holy grail is in predicting protein conformational dynamics, not just static structures
It turns out that proteins are usually thought of as static arrangements of atoms in space; however, biologists know very well that protein function actually depends on how they move -what they dub “dynamics”, “flexibility”, or, when referring to distinct structurally stable states that interconvert, “structural” or “conformational” diversity.
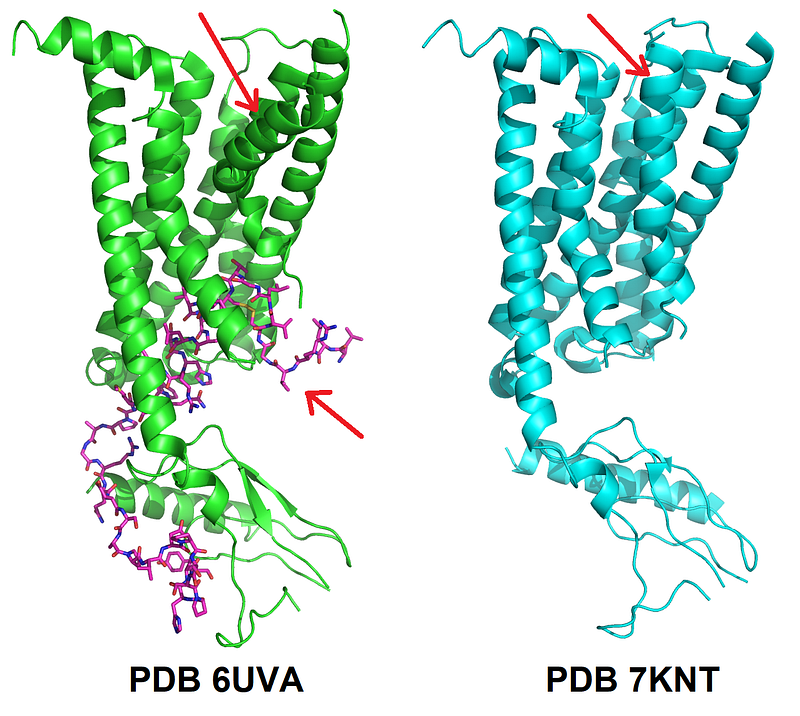
Typically, experimental structures only capture specific conformational states, or in the best case a few states drawn from a larger pool of possible conformations and stabilized in some way. Since methods for protein structure prediction are trained with this biased data, they tend to predict certain states over others that are less represented in structural databases. In general terms, AlphaFold doesn’t escape from this limitation.
However, for one of the CASP14 targets, AlphaFold 2 modeled multiple conformations, two of which were consistent with certain sets of data. This was a membrane protein whose function is to transport small molecules across the membrane. The job of this kind of proteins, called transporters, is to take a molecule on one side of the membrane and release it on the other. To do this, transporters need to undergo important structural changes. AF2 had in CASP14 captured such changes when predicting the structure of a transporter. Then the question was obvious: can we infer such structural changes more broadly? The paper by the Mchaourab and Meiler groups (del Alamo et al eLife 2022;11:e75751, see direct link at the end) tackles precisely this.
The work tested AF2’s capacity to model the multiple states of various transmembrane proteins whose structures had not been used to train AF2, as they were released into the public domain after CASP14. The conclusion is that there are indeed ways to tweak AF2 runs to produce structural heterogeneity in meaningful ways. Although no exact generalization protocol is yet available, this is the main rationale behind it:
A regular AF2 run starts with a query to a database of protein sequences to generate a multiple sequence alignment (MSA). A randomly sampled subset of this MSA is then fed into AF2's main neural network three times, to produce several 3D models. When MSAs are “deep” enough, meaning they contain sufficiently large numbers of sequences, the procedure typically converges in such a way that all the obtained models are very similar. What del Alamo et al observed is that restricting the depth of the input MSA results in AlphaFold2 producing structurally more varied models, and that one could fine-tune how much of the original MSA to feed the neural network in order to produce meaningful structural variation.
The setting is not straightforward, because too small or poorly curated alignments can simply result in artifacts and even regions that look flexible simply because they cannot be properly defined with the available data; while on the other hand too much data will most likely end up shifting all models to the same structure. At least, the work is important because it validates a relatively simple approach to one of the biggest problems in structural biology. In fact, in my article above I explain how CASP (the competition that Deepmind won with its AlphaFold programs) is going to dedicate part of its next edition to predicting structural variability -which just didn’t make sense before AlphaFold showed up.
As a structural biologist I can nothing but hope that the hype on structure prediction will not stop here.
References
The original paper:
An insightful comment on the paper:
Outreach articles I wrote on AlphaFold, CASP, and protein structure prediction:
Are you interested in doing a project with me about protein modeling, bioinformatics, protein design, molecular modeling, or protein biotechnology? Contact me here!
www.lucianoabriata.com I write and photoshoot about everything that lies in my broad sphere of interests: nature, science, technology, programming, etc. Become a Medium member to access all its stories (affiliate links of the platform for which I get small revenues without cost to you) and subscribe to get my new stories by email. To consult about small jobs check my services page here. You can contact me here.