Tutorial on Density Functional Theory using GAMESS
This tutorial is for beginners who are interested in learning how to set up and run a first-principle calculation based on density functional theory (DFT). DFT is the most widely used method by quantum chemists, condensed matter physicists, and material scientists for calculating important materials properties such as equilibrium geometry, quantum energy levels, optoelectronic properties, vibrational properties, IR spectrum, etc.
DFT can be used for both molecular systems (finite size) or extended periodic systems like solids. In a previous article, we discussed how to set up and run a DFT calculation for a periodic 2D layered system: Tutorial on Density Functional Theory using quantum espresso. For DFT studies of 1D nanomaterials such as carbon nanotubes and graphene nanoribbons, see the following: Tutorial on DFT Studies of 1D Nanomaterials Using Quantum Espresso.
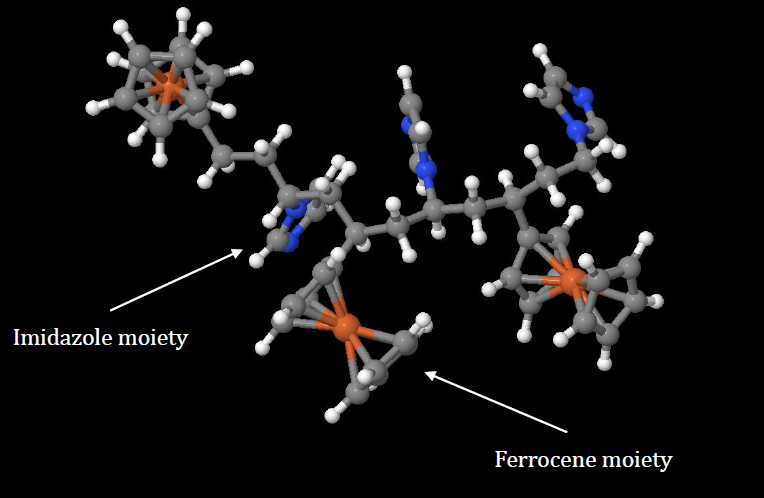
Here we focus on DFT calculations for a molecular or polymeric system. As an example, we consider an organometallic copolymer containing ferrocene and imidazole, as shown in the figure above.
In this tutorial, we consider a geometry optimization calculation only. An excited state calculation can be performed on the converged equilibrium geometry using time-depending density functional theory (TDDFT) to obtain excited state properties such as excitation energies and transition probabilities (oscillator strength). We will discuss TDDFT calculations in a future article.
All calculations are performed using the GAMESS DFT Solver. For more information, see the following link: https://www.msg.chem.iastate.edu/gamess/.
Building the Initial Geometry of the Polymer
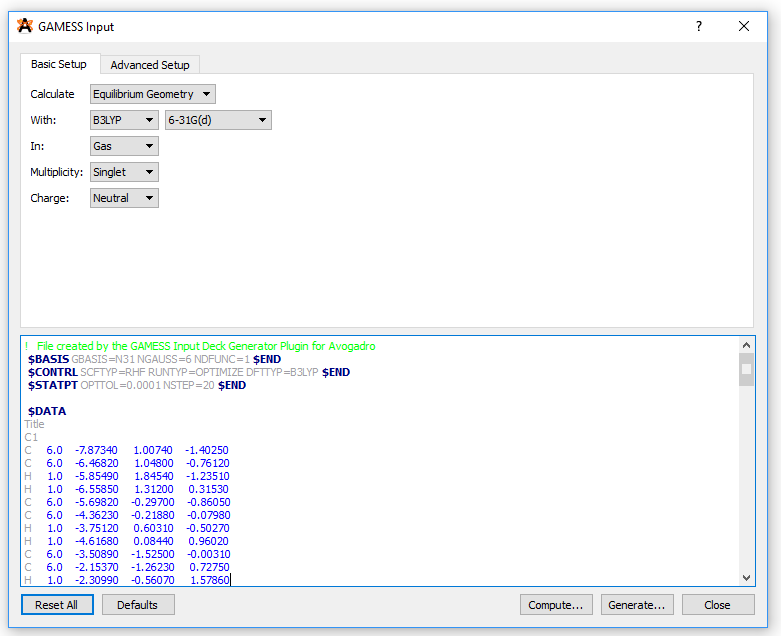
Molecular geometries of the polymers can be created using the Build Polymer tool in the Maestro program or using the Avogadro software. After building the initial geometry, the geometry should be stored in an xyz file format containing xyz coordinates of all atoms in the polymer. Then import the xyz file into Avogadro. Once imported, click on Extensions -> GAMESS -> Input Generator. This will open a new window where you can create your input file for running a GAMESS calculation:
Run a Geometry Optimization Calculation
The final input file (input.inp) generated from Avogadro can submitted to a cluster. Note that under the $CONTRL key in the input file, we selected RUNTYP=OPTIMIZE since we are running a geometry optimization calculation. A simple batch script (input.sl) for running the calculation would look like this:
#!/bin/bash -l
#SBATCH -J test_gamess
#SBATCH -q regular
#SBATCH -N 1
#SBATCH -C haswell
#SBATCH -t 48:00:00
#SBATCH [email protected]
#SBATCH --mail-type=ALLcd $SLURM_SUBMIT_DIR
module load gamessrungms-xt input.inp 32To submit the job, use the following command:
$ sbatch input.slAnalyzing Results from Geometry Optimization Calculation
A geometry optimization calculation provides useful information such as:
- Geometrical parameters (bond lengths, bond angles, dihedral angles).
- Frontier energy levels (HOMO and LUMO energies).
- Converged electronic density which can be used for visualization of molecular orbitals.
- Vibrational frequencies and thermodynamic properties if RUNTPY = HESSIAN.
- UV-Vis spectrum if RUNTYP=ENERGY and TDDFT=EXCITE is requested, that is a TDDFT calculation. For a TDDFT calculation, the input file would look like this:
! File created by the GAMESS Input Deck Generator Plugin for Avogadro
$BASIS GBASIS=SBK NGAUSS=3 NDFUNC=1 $END
$CONTRL SCFTYP=RHF RUNTYP=ENERGY DFTTYP=B3LYP TDDFT=EXCITE ECP=SBK $END
$CONTRL MAXIT = 200 $END
$SCF CONV=1.0d-06 $END
$TDDFT NSTATE=20 $END
$SYSTEM MWORDS=2400 MEMDDI=1600 $END$DATA
Title
C1
C 6.0 -7.8733754917 1.0074296340 -1.4025054267
C 6.0 -6.4681676425 1.0479963249 -0.7611564649
H 1.0 -5.8548660623 1.8454068507 -1.2350812070
H 1.0 -6.5585148572 1.3119926132 0.3152674370
C 6.0 -5.6981500150 -0.2970003447 -0.8605182087
C 6.0 -4.3623058083 -0.2187969941 -0.0797789426
H 1.0 -3.7511581242 0.6031219603 -0.5026731629
H 1.0 -4.6168279837 0.0844225852 0.9601947003
. . . . .
. . . . .
. . . . .
$ENDGAMESS’ most closely integrated graphical interface is MacMolPlt, which is now available for Mac OS X, Linux, and Windows desktop platforms. MacMolPlt is extremely useful for showing results from the output files of GAMESS. Other softwares that could be used for visualizing the output file from a GAMESS calculation are Avogadro and jmol.
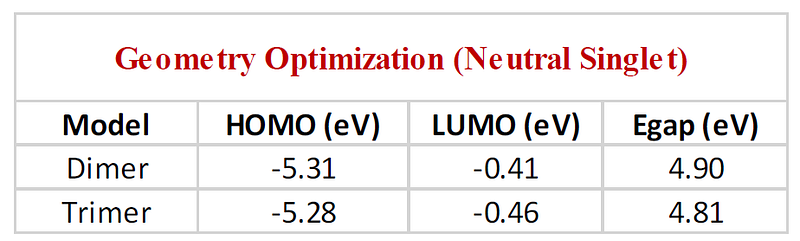
For the polymer under consideration, a sample output for the HOMO and LUMO energies is shown in the table below:
Molecular orbitals of interest (requires knowledge from TDDFT calculations for determination of oscillator strengths)
HOMO Plot
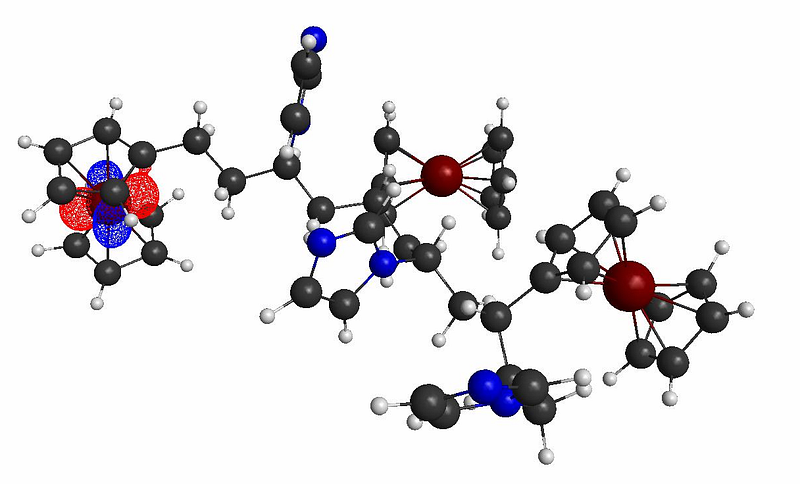
LUMO+6 Plot
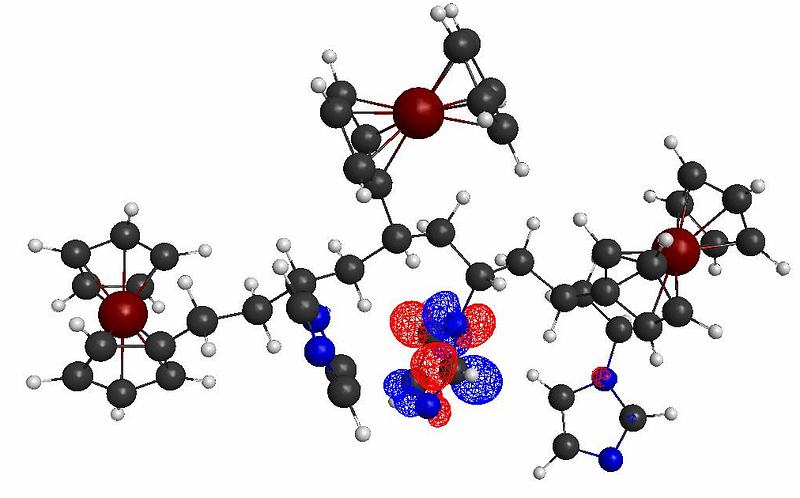
The molecular orbital plots indicate the formation of possible donor/acceptor complexes within the polymer, with the Imizadole acting as the acceptor molecule (due to its strong electron affinity), and the ferrocene acting as the donor molecule. This polymer thus show strong potentials for photocurrent generation and photoconductivity applications, as shown below:
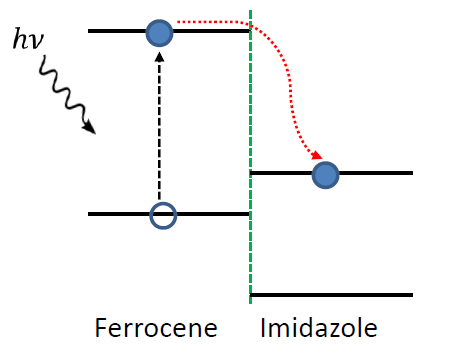
Key Points to Keep in Mind When Running a Geometry Optimization Calculation
Always ensure the SCF solution for geometry optimization does not converge to an excited state (higher local minima) which can lead to misleading results. To ensure the geometry converges to a global minimum, you may have to reduce the convergence criterion to something like 0.0001 Hatree/Bohr. Also try different combinations of exchange-correlation models and basis sets, and find out what works best for your system. For more information, see the following article: “Comments on the Molecular Geometry of Ferrocene: The Dangers of Using Quantum Chemistry Programs as Black Boxes”, J Comput Chem 30: 881–883, 2009. Here is table from the article:
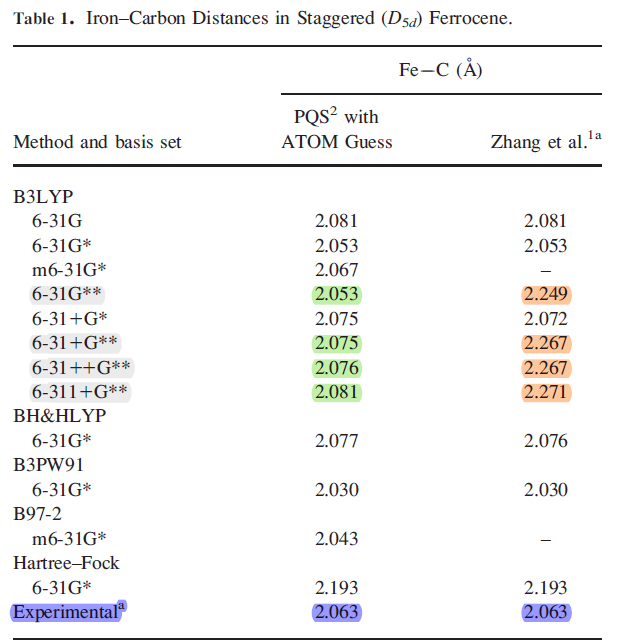
The table shows clearly that DFT calculations could yield longer bond distances that strongly disagree with experiment if the calculation fails to converge to a global minimum.
In summary, we have provided a tutorial that can help beginners to set up and run a DFT calculation for molecular or polymeric systems. For more information on how to create various input files as well as tuning different hyperparameters such as convergence criteria, see the GAMESS input file description documentation which can be found here: https://www.msg.chem.iastate.edu/gamess/documentation.html.
References
- GAMESS URL: https://www.msg.chem.iastate.edu/GAMESS/GAMESS.html.
- NERSC for supercomputing resources: https://www.nersc.gov/.





