
AI-first Biotech
The Road to Biology 2.0 Will Pass Through Black-Box Data
This year marks perhaps the zenith of expectations for AI-based breakthroughs in biology, transforming it into an engineering discipline that is programmable, predictable, and replicable. Drawing insights from AI breakthroughs in perception, natural language, and protein structure prediction, we endeavour to pinpoint the characteristics of biological problems that are most conducive to being solved by AI techniques. Subsequently, we delineate three conceptual generations of bio AI approaches in the biotech industry and contend that the most significant future breakthrough will arise from the transition away from traditional “white-box” data, understandable by humans, to novel high-throughput, low-cost AI-specific “black-box” data modalities developed in tandem with appropriate computational methods.

This post was co-authored with Luca Naef.
The release of ChatGPT by OpenAI in November 2022 has thrust Artificial Intelligence into the global public spotlight [1]. It likely marked the first instance where even people far from the field realised that AI is imminently and rapidly altering the very foundations of how humans will work in the near future [2]. A year down the road, once the limitations of ChatGPT and similar systems have become better understood [3], the initial doom predictions ranging from the more habitual panic about future massive job replacement by AI to declaring OpenAI as the bane of Google, have given place to impatience — “why is it so slow?”, in the words of Sam Altman, the CEO of OpenAI [4]. Familiarity breeds contempt, as the saying goes.
We are now seeing the same frenetic optimism around AI in the biological sciences, with hopes that are probably best summarised by DeepMind CEO Demis Hassabis’ declaration that AI will be used to “solve” biology [5]. At the same time, we must admit that AI revolutions in biology have been surprisingly limited in scope. Indeed, the largest real revolution so far has been in protein structure prediction, with DeepMind’s AlphaFold [6] already transforming many workflows, with many questions completely unthinkable just a few years ago now being common practice [7]. Similarly to ChatGPT, as the capabilities and limitations of AlphaFold become better understood [8], the initial shock of “AI will replace structural biologists” has turned into “why is it so slow?”
“Who traces life and seeks to give
Descriptions of the things that live
Begins with killing to dissect
He gets the pieces to inspect
The lifeless limbs beneath his knife
All parts — but link that gave them life!”
— Goethe, “Faust”
One of the biggest challenges of biology is the vast number of temporal and spatial scales, from the nanometer (10ᐨ⁹) scale of molecules to the centimetre (10ᐨ²) scale of tissues and organs, and from femtosecond (10ᐨ¹⁵) scale of atomic interactions to years (10⁷) of a typical organism lifetime. It is often the case that while we have a very good understanding of the laws governing the smaller scales, e.g., the Schrödinger equation describing the quantum mechanical system of atoms comprising a protein molecule, we cannot simply extend them to the larger scales due to computational intractability [9]. At the same time, the laws operating at the larger scales are much more elusive and do not directly derive from the lower-level ones [10]. One of the big hopes for AI in life sciences is to provide a data-driven way to bridge these scales, possibly ushering a new age in science that was dubbed “post-theory” [11]: AlphaFold may produce accurate predictions of protein fold structure, but it is a black box that does not offer a theory of protein folding, or at least not one that a human scientist can understand [12].
AI revolutions of yesteryears
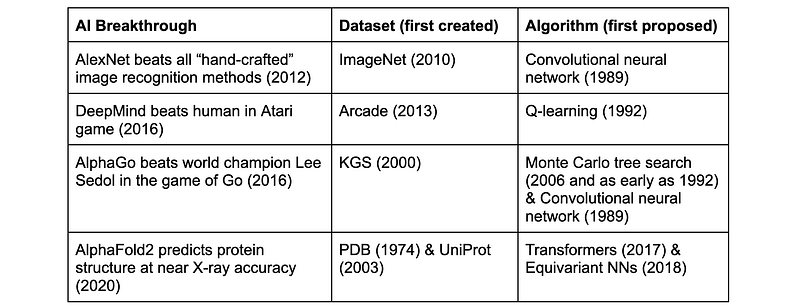
Previous AI breakthroughs, in particular the rise of deep learning in perception and language over the past decade, have been primarily attributed to the confluence of three factors: new models, more computational power, and more data. It is the latter that has arguably been the most crucial component of success, captured in the laconic maxim “data is king.” The major success of deep learning in computer vision in 2012 [13] was enabled due to the availability of large-scale training data — millions of labelled images of the ImageNet benchmark introduced in 2010 [14]. While the ML model itself was known for nearly two decades [15], it took only three years from the publication of the dataset to the breakthrough. A similar deep learning revolution happened in natural language processing, where significant gains were achieved with old models [16] on newly introduced large-scale benchmarks [17]. More recent progress in these fields has been primarily driven by scaling laws both in the computing power and available data [18].
Interestingly, with the AlphaFold2 2020 breakthrough, the situation seems to be reversed: the data used for its training (protein structures and sequences) existed for decades and was of modest scale [19], whereas the ML model was novel and recent: it explored the 2017 Transformer architecture together with geometric invariance ideas that have become popular around the same time [20]. AlphaFold thus breaks away from the previous trend of “datasets over algorithms” [21] and raises another crucial but often overlooked success factor: the right choice of the problem.
Why did AlphaFold work?
In his 1972 Nobel lecture, Christian Anfinsen postulated that a protein structure is fully determined by its amino acid sequence [22]. It turned out to be easy to say but hard to do, and the protein folding problem became the Holy Grail of structural biology for the following fifty years. A biannual competition, the Critical Assessment of Structure Prediction (CASP), set up in 1994, tracked the progress in this notoriously difficult problem by comparing the accuracy of predictions against novel experimentally-determined structures, unreleased to the public.
In 2018, the biological community experienced an “ImageNet moment” when DeepMind outperformed by a large margin all the competitive techniques with the first version of AlphaFold [23]. In 2020, AlphaFold2 reached structure prediction accuracy “comparable” to that of X-ray crystallography, heralding a new era in protein science.
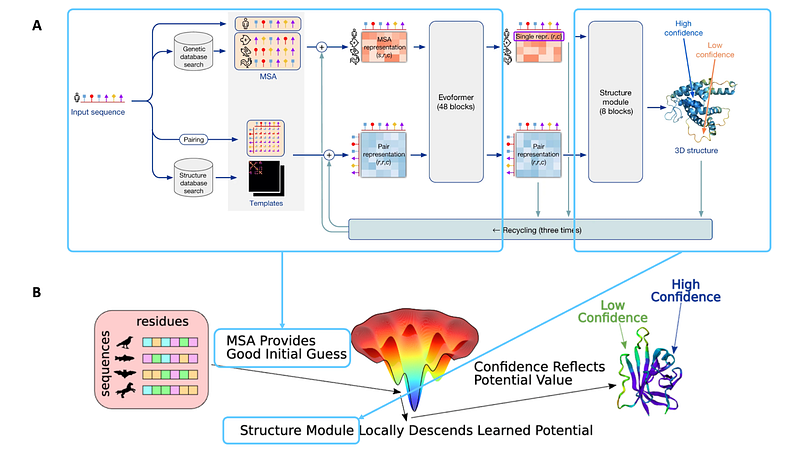
The AlphaFold model relied both on structural information as well as the evolutionary history of proteins, provided in the form of multiple sequence alignment (MSA). In 2022, a Meta team (now a startup Evolutionary Scale) published a method capable of predicting protein structure from a single sequence called ESMFold [24], based on the AlphaFold2 architecture and replacing evolutionary information extracted from the MSA with embeddings derived from a Large Language Model (LLM), ESM2.
Unusually good training data. Trying to understand the anatomy of AlphaFold2 in an attempt to replicate its success in other applications of biology, it is natural to start with perhaps the most crucial ingredient of a deep learning model: the data on which it is trained. Protein structure prediction appears to be rather unique due to its uncharacteristically homogenous and large-scale (by biology standards) structural data collected with X-ray crystallography or cryogenic electron microscopy (cryo-EM) technologies that come in the forms of estimated absolute atomic spatial coordinates.
This is contrasted to the often “relative” quantities in other fields of biology, such as RNA sequencing, mass spectrometry, or cell painting, which are often plagued by strong “batch effects’’ and readout being relative to “background noise”, making, even after normalisation, different experimental setups hard to compare, leading to fewer successes of building foundation models in these areas [26]. Combining data sources for many of these modalities is still a very active area of research [27–28].
Many other approaches also suffer from low dynamic range, where some signals (e.g. certain highly-expressed genes) are so abundant that signals from less abundant genes get drowned in background noise. Protein structures are thus unique in being unusually homogenous, diverse, and absolute data (3D coordinates in space), with minimal deviations between different labs resolving the same structure.
“Degenerate” solution space. Another peculiarity of AlphaFold2 is that the supervised training set of only 140K protein structures and 350K sequences and is tiny by ML standards [29] — an order of magnitude less than the amount of data used to train AlexNet almost a decade earlier, and a drop in the ocean compared to the contemporaneous GPT-3 [18]. What likely makes such a small dataset sufficient is the “degeneracy” of the solution space: while in theory the number of all possible solutions in protein folding is astronomically large (estimated at 10³⁰⁰ [30]), only a very small fraction thereof is actualised. This is akin to the “manifold hypothesis” in computer vision, stating that natural images form a low-dimensional subspace in the space of all possible pixel colours [31].
The reason for this “degeneracy” likely lies with evolution: most of the proteins we know have emerged over 3.5 billion years of evolutionary optimisation in which existing domains were copied, pasted, and mutated [32], producing a limited “vocabulary” that is reused over and over again. There are thermodynamic reasons for this, too, as only a limited set of possible amino acid 3D arrangements make up for the entropic cost of a defined protein fold [33]. Most protein folds can thus be achieved by recombining and slightly modifying existing ones and valid solutions can be formed through advanced retrieval techniques [34].
From this perspective, the protein folding problem is reminiscent of natural language, where tasks such as writing, coding, translation, and conversation where LLMs excel and which often do not require strong generalisation and can be solved by recombining existing examples [35] (e.g. copy-pasting pieces of code from GitHub code repositories that GPT-4 has been trained on).
Thus, while AlphaFold2 has worked remarkably well in predicting the structures of proteins never crystallised before, it might be not due to its generalisation capabilities but on the contrary, because it does not need to generalise [36]. An observation in favour of this hypothesis is that even the most recent update to AlphaFold (AlphaFold-latest, which introduces small molecules), and other state-of-the-art docking algorithms appear to struggle in generalising to previously unseen protein-ligand complexes [37–38], as unlike proteins most small molecules do not evolve, and therefore the solution space is likely less degenerate and less well reflected in the training data [39–40].
“Distributional hypothesis.” Another key parallel between language-related tasks and protein structure prediction is the “distributional hypothesis”, which says that words occurring in the same contexts tend to have similar meanings [41]. In human language, much of the meaning of a word is derived from its context. For example, depending on the context, the word “bank” can mean a financial institution or the side of a river. The masked language modelling and next token prediction objectives employed in today’s LLMs, and also used in AlphaFold2 and ESMFold, works by masking out a set of words (more correctly, “tokens”) and having the neural network reconstruct them [42]. Due to the semantic structure of human language, this simple task allows the model to learn the fundamental semantic meaning of words and phrases. And since the solution space is degenerate, most problems can be solved by just infilling or extending with these most likely sets of words.
In proteins, the relative placement of domains within a protein and amino acids within a domain are mostly pre-determined by the protein’s function [43]. Since AlphaFold2 is trained and evaluated primarily on functional proteins (products of successful evolutionary optimisation), it is provided with an extremely strong signal to learn the “distributional hypothesis” for protein sequences and structures. Roughly speaking, to fold new sequences, AlphaFold2 can just “retrieve” a rough set of global domain or motif arrangements, and then connect them. AlphaFold2 achieves this by using the evolutionary history of proteins through MSA and ESMFold through a pre-trained LLM. This is evident from the strong dependence of AlphaFold on the MSA depth [6] and that of ESMFold on the number of similar neighbours in the training data [24], its failure to reliably predict multiple stable conformers for fold-switching proteins [44–45], and the fact that both AlphaFold and ESMFold tend to be insensitive towards isoforms that have large deletions and are unlikely to fold into the same state as the native proteins [40].
Clearly defined performance criteria. One of the reasons for the rapid progress of deep learning in perception and language has been the existence of well-defined and broadly accepted benchmarks, comprising datasets used for training and testing and an evaluation protocol — e.g., ImageNet in computer vision and GLUE in natural language processing. For protein structure prediction, such a benchmark was CASP. Comparing predicted 3D structures to the experimentally-determined groundtruth is also straightforward using root-mean-square distance (RMSD), leading to a clearly defined loss function that is crucial for the success of deep learning-based methods [4].
We can conclude that there are good reasons for AlphaFold to stand out as perhaps the most famous isolated breakthrough in biological AI, thanks to the nature of both the data (protein structures and sequences are unusually uniform and high-quality compared to other types of biological data) and the problem (while theoretically extremely high-complexity, the protein folding problem seems to benefit from a very degenerate solution space). As a result, as suggested by a recent slew of results [25, 44–45, 47], one can likely solve protein structure prediction without having to learn any physics. In other biological problems such as molecular property prediction, target-disease associations, and even protein-ligand docking, the picture might be much less optimistic.
Is there a recipe for bioAI success?
AlphaFold2 is a convincing example that ML can work for the right kind of biological problems in the right setting. It is also the beginning of a path towards “Biology 2.0” [49] as proclaimed by Nvidia CEO Jensen Huang [50]: applying engineering and computer science principles and tools to biological problems, bringing the physical and digital worlds together, with the purpose of both gaining understanding of biology and manipulating it (e.g., for drug development). Learnings from AlphaFold as well as from analogies to previous AI breakthroughs in language and perception might help better direct investment of efforts and capital toward Biology 2.0 and avoid the “trough of disillusionment” in biological AI.
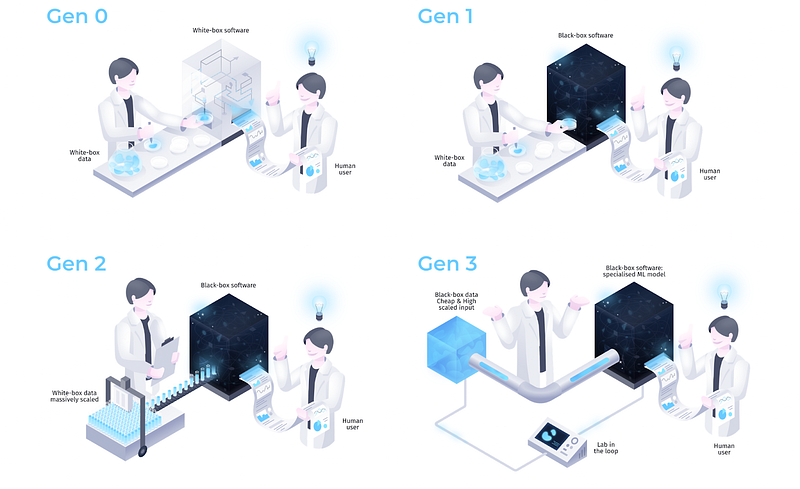
For this purpose, we find it illuminating to closely examine the conceptual evolution of biological AI approaches. Since many of these efforts have happened in the biotech industry, we will focus on various generations of AI-first companies, which we define as follows [51]: “Generation 1” companies apply ML to existing data modalities, which are not necessarily a good fit. For this reason, Generation 1 companies have so far shown limited success compared to traditional drug discovery approaches. Whenever people talk about AI in biology being “over-hyped”, it is usually these “Generation 1” companies they have in mind.
“Generation 2” companies produce data in a systematic way by scaling existing experimental technologies. We believe a subset of these focusing on the right problems and having the right data generation capabilities will succeed in the long run.
“Generation 3” companies develop purpose-built new (or repurposed old) experimental techniques enabling the production of ML-specific data that focuses on problems with a “degenerate solution space”. We expect the biggest breakthroughs in AI-based drug discovery to come from this co-development of ML and experimental technology, and more generally, see it as a necessary step toward the future AI-based biological sciences.
Generation 1: new ML methods for old data
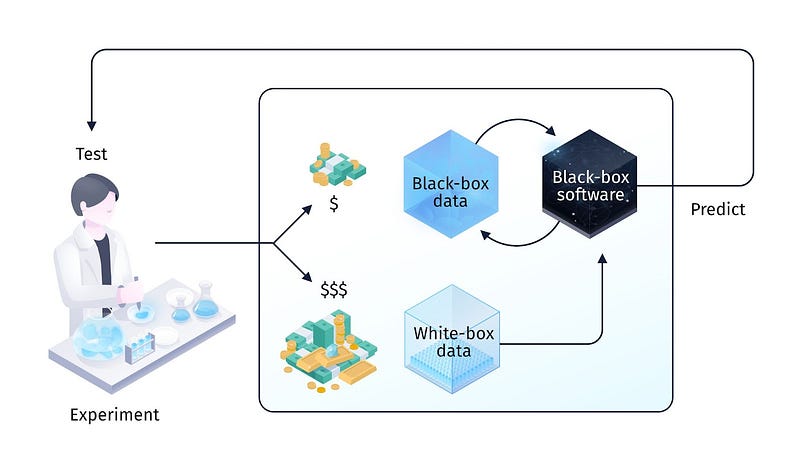
In the traditional biotech industry (“Generation 0”), the data was collected by human scientists for human scientists and was analysed using human-designed algorithms based on their best understanding of the problem. We call this classical paradigm “white-box data / white-box software.” With the advent of deep learning algorithms, multiple biotech companies attempted to expand their groundbreaking successes in perception and language into biological problems in order to discover and then drug therapeutic targets. The key difference between the use of deep learning and traditional computational methods is perhaps best captured by the term “software 2.0” coined by Andrej Karapathy [52]. In software 1.0, which spanned from the early days of the ENIAC to present-day computers, software algorithms were designed by human ingenuity, first deriving and then programming the underlying logic into instructions the computer can understand. After programming, Software 1.0 is then fed with data to produce the desired behavior.
Software 2.0 is fundamentally different: algorithms are programmed not by feeding instructions but by feeding data, and the desired behaviour (in the form of a loss function) from which neural networks then extract the right logic to solve the problem on their own — given enough compute and a suitable optimisation algorithm that maximises the desired behavior. The logic is stored implicitly in the neural network weights. The resulting algorithm acts as a “black box” without humans being able to fully understand these implicit instructions. While “black box” often has a negative connotation, it was this shift from hand-crafted features and instructions to model-extracted “black-box” features and instructions that was one of the defining characteristics and reasons for the success of deep learning models.

Generation 1 emerged in the early 2010s with the idea of applying deep learning methods (“black-box software”) to existing “white-box” biological data. Prominent bio AI companies of this generation, Atomwise and Exscientia (both founded in 2012), attempted to accelerate hit discovery by replacing traditional protein-ligand docking and screening techniques with ML-based ones. The protein-ligand docking problem, however, as opposed to protein folding, does not have important characteristics that make the latter problem tractable. First, the data is extremely skewed and significantly less diverse than PDB. The vast majority of ligands are small molecules that are easy to synthesise and the majority of targets are proteins that are easy to crystallise or highly biassed towards pharmaceutically exciting targets (e.g., kinases); medicinal chemists have the least problem with drugging them. Second, the solution space for ligands is not degenerate, since, unlike proteins, small molecules are generated not through evolution but through chemical synthesis [54, 39].
Consequently, small molecules lack the distributional hypothesis we see in proteins: the positions of building blocks within a molecule are often explained not by function but by what is possible to synthesise with standard chemical processes. Masked language modelling or next token prediction on the existing chemical matter thus is most likely to learn synthesis biases rather than function. This is seen in the lacklustre performance of LLMs for small molecules trained on databases like Pubchem and ChEMBL, compared to simple non-deep learning baselines [55]. More generally speaking, ML-based protein-ligand docking and molecular property prediction approaches manifest poor performance when evaluated on novel chemical or protein data, exactly the setting where good generalisation capabilities are direly needed.
Similar challenges exist with disease-target associations, the early focus of another prominent Generation 1 company Benevolent AI (founded in 2013), which initially tried to use NLP approaches to mine scientific literature. While the data in principle could be substantial, it is extremely hard to determine whether a model predicted a novel disease association, since one can find associations for almost any protein with almost any disease in the literature. Due to inconsistency in nomenclature, it is nearly impossible to enssure that any newly discovered links are actually novel. On top of that, the use of academic literature as data source turned out to be extremely noisy, as some published results may be false or irreproducible [56]. In addition to inadequate data, the solution space might also not be degenerate: many complex diseases have very diverse and elusive mechanisms, so the hope to “copy-paste” known solutions with small changes might be wishful thinking.
On top of that, Generation 1 companies face an increasingly challenging business model. Curation of public datasets is no longer a competitive advantage, and with the emergence of ML frameworks such as PyTorch, deep learning algorithms, once a significant competitive edge requiring very specialised knowledge of an exclusive cohort of scientists, have been dramatically democratised and are now accessible to an undergraduate student. Scale still constitutes a competitive advantage as few companies have appropriate computational resources, but not an unbridgeable one. In short, being a Generation 1 company is not a defensible competitive advantage anymore (and most likely, has never been [57]). For these reasons, combined with less forgiving capital markets over the past two years, many Generation 1 companies have already pivoted into a classical pharma model focusing on their existing clinical assets or were forced to undergo downscaling and restructuring, and likely many more will.
Generation 2: scaling old data generation modalities
Responding to these challenges, a new class of companies emerged towards the mid-2010s, combining ML with large-scale biological data generation modalities to address in particular the aforementioned problems with existing data. These companies, which we call “Generation 2,” still fall under the “white-box data / black-box software” concept, with the important difference being the ability of Generation 2 companies to generate their own data at scale suitable for ML [58]. Prominent Generation 2 companies Recursion (founded in 2013) and Insitro (2018) initial claim to fame was the scaling of cell painting technologies to produce hundreds of millions of cell phenotype images. In this technology, cell phenotypes are captured across multiple channels with multiple fluorescent dyes, visualising cellular components such as the nucleus, endoplasmic reticulum, cytoplasmic RNA, and mitochondria, which can then be used in the drug discovery context to predict bioactivity, toxicity, and understand mechanisms of action of chemical and genetic perturbations [59]. Generate Biomedicines (2018) has developed perhaps one of the largest cryo-EM facilities to acquire protein structures. Insitro, Zebi AI, and Anagenex (all 2019) have developed large-scale DNA-encoded library (DEL) screens that measure protein binding of massive amounts of molecules tagged with unique DNA “barcodes” against a single drug target.
DEL-approaches in particular, also fulfill the “degeneracy” hallmark. With ultra-large (up to billions) screening libraries, DEL approaches produce a large number of hits (“solutions”) against the screened target, which, however, are often large and not drug-like molecules. DELs thus lead to “degenerate solution spaces” against individual targets, in the sense that first, a large number of hits are produced experimentally and subsequently ML is used to extract patterns from these hits to derive novel solutions. This is a fruitful strategy as “copy-paste with small changes’’ then becomes a viable path for the ML methods to produce more drug-like hits and leads, and this is where current ML approaches excel [60]. However, DELs are unlikely to transform protein-ligand interaction discovery against completely novel, hit-free targets to a “degenerate solution space,” as they do not scale to many proteins and there is no generalisable (e.g., structural) information that is obtained.
Generation 3: co-development of ML with novel biological data acquisition technologies
While it is likely that some of these Generation 2 companies will be successful, and their approaches have strong scientific merit compared to Generation 1 companies, we believe that there will be a third generation that leverages ML even more successfully. This is because Generation 2 companies are still bound by the disadvantages of “white-box data” modalities that were originally developed to be used by human intelligence and in most cases emphasise quality over quantity. There are good reasons for this historically, as biology is a signal-to-noise problem, and false positives (“type I error”) tend to be the most expensive mistakes since they lead to chasing down the wrong hypothesis with very expensive experiments.
Traditional biological assays thus trade high throughput for high precision. This, however, is not the right strategy for artificial intelligence, which is much better at handling noise and where models are often pre-trained on very large, diverse, yet noisy, datasets (such as CommonCrawl, text data crawled from the internet), and fine-tuned on a small set of high-quality data points (e.g., via Reinforcement Learning from Human Feedback, successfully used in ChatGPT).
Furthermore, biological sciences tend to suffer from the lack of true negatives, which is partly a consequence of the academic incentive and publication model: an unsuccessful experiment is rarely reported in the scientific literature, its data is discarded, and a human scientist goes back to the drawing board. However, such data is extremely useful for AI, and the ability to produce large amounts of negative examples is at least as important as having many positive ones.
We believe that the scarcity of data and the cost of its acquisition using existing modalities focused on minimising false positives as well as true negatives constitute a fundamental bottleneck towards AI-driven “Biology 2.0.” For example, it took over two decades for cryo-EM, currently one of the two main modalities used for imaging protein structure, to improve resolution by an order of magnitude (from approximately 20Å to 2Å [61]) and decrease the cost approximately two-fold. At the current price point (of the order >$10k per typical protein), obtaining the cryo-EM structures of all proteins with known sequences would easily cost in the range of trillions of dollars. Data scarcity is, in our view, one of the reasons why we have so far only seen a limited biological impact of algorithmic advances in generative AI that in the past few years have achieved remarkable performance in extremely data-rich domains such as language and image generation [62].
Hence, we believe that the most exciting direction for further investment is completely novel low-cost high-throughput biological data modalities that may appear as “black-box” to humans. They will explicitly trade interpretability for quantity and will not just be scaled-up data optimised for human hypothesis evaluation, but data specifically designed to train machine learning systems that will then produce the required human-understandable output for hypothesis evaluation. We call this new paradigm “black-box data / black-box software” and believe it will be the core of new Generation 3 biological AI companies [63].
From an epistemological perspective, the Generation 3 mindset challenges the traditional scientific discovery paradigm. In the traditional hypothesise–predict–test loop that has been at the foundation of the scientific method for centuries, white-box experimental data is used by a human scientist to propose a hypothesis, which is then used to make predictions and test them experimentally in order to confirm or reject the hypothesis [64]. In Generation 3 approaches, hypothesis generation is done by ML models trained on black-box data, and then tested on white-box data. It is this decoupling of the data from the requirement for human understanding that can potentially unlock massively larger scale and diversity of experimental technologies [65].
Crucially, black-box data is still collected from the original sample under question (e.g., a protein structure in case of protein folding, or an actual human in case of human phenotype) rather than a simplified model system thereof, thus maintaining full fidelity [66]. Black-box data in this case is just another data source from the same system that is significantly easier to obtain than the white-box data but still informative of the downstream prediction. The bigger the difference in obtaining white- vs black-box data for such a sample, the more impactful the use of the latter is likely to be. Well-known example are biomarkers or surrogates to predict a human phenotype (e.g., the levels of apolipoprotein Apo-B100 for Atherosclerotic Cardiovascular Disease). It also makes clear the requirement to have sufficient paired “white-box” + “black-box” data to train and evaluate such a model.
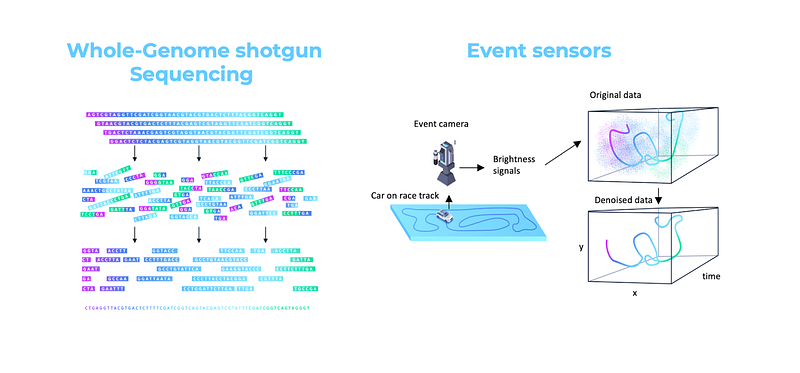
An analogy from the domain of imaging sciences is computational photography. Traditional digital imaging systems developed towards the end of the last century operated with CCD or CMOS sensors producing image data that looked like the desired output picture with certain imperfections, noise, or artifacts (hence “white-box data”), that were reduced or removed by appropriately designed image processing algorithms (“white-box software”). The success of deep learning in the past decade has replaced hand-crafted “white-box” image processing algorithms with “black-box” neural network-based filters. Modern imaging systems such as those found in Apple iPhone go further, replacing the human-intelligible data with inputs from multiple sensors or modalities that do not necessarily look like the output picture (hence “black-box data”) and fusing them with the help of deep learning. Some extreme examples of such “black-box data modalities” include event cameras capturing pixel brightness differences at megahertz rates [67] or compressed sensing, in which the inputs are carefully designed random projections [68].
Even within the realm of biology, unlocking new insights by moving away from human-processable data is far from new. Trading off some data interpretability for an exponential increase in scale has been the key to probably one of the biggest achievements in biological sciences to date — the Human Genome Project. Sequencing the human genome was made possible in large parts through “whole-genome shotgun sequencing,” a technology developed by Craig Venter and his company Celera, which exactly employs this strategy. Moving away from manual painstaking piece-by-piece assembly of the genome to smashing it into millions of human-unintelligible pieces that were then re-assembled using alignment algorithms allowed to generate exponentially more data, which has led to Celera eventually winning the “genome war” [69]. Deep learning was not used at that time, so this example is what we would call “black-box data / white-box software.”
We believe that the new Generation 3 methods are our best shot at systematically mapping biology to generate the data needed to feed Software 2.0 and to train foundational ML models beyond those available today. This represents a fundamental paradigm shift, as it will be data that has no meaning for humans directly but will gain value when combined with ML and generated only for this purpose. It means moving away from optimising from specific human inquiry to systematic, AI-focused experimental methods.
This is a shift that is unlikely to come from traditional academic institutions, since life scientists are more interested in discovering novel things than building novel tools, and the new mindset requires large, long-term, interdisciplinary, coordinated efforts that do not fit into the PhD or tenure model. Likewise, it is unlikely to come from the classical pharmaceutical industry, as it does not directly tie into drug discovery programs or fit within the pharma business model centred around disease areas. Probably, the shift will come from Generation 3 biotech startups or novel AI-first academic bio labs [70], each placing a “big bet” on co-developing a novel data modality and corresponding ML algorithms. And like only the combination of training on the noisy swaths of the internet together with small datasets of hand-curated human feedback enabled ChatGPT, they will work in conjunction with a smaller set of existing, high-quality, and low-throughput human interpretable data.
We foresee that the transition into “Generation 3” will take the form of extending the complexity of the tasks while using existing data sources (such that the data does not anymore directly contain sufficient information for a human about the task it is collected for), repurposing known experimental technology that has not been considered appropriate for particular tasks, and finally, coming up with completely novel data acquisition modalities. The first shift is already happening in in companies that initially were founded as “Generation 2.” One such example is Recursion, which has recently announced it will move from cell-painting technology on which it was founded, to bright-field imaging that does not require any staining (hence cheaper and faster to acquire) and allows to image live cells over time and possibly used causal models [71].
We also see instances of repurposing known or developing new experimental technology in several recently founded “Generation 3” companies, such as chemical mapping developed by Atomic AI (founded in 2021) for RNA-targeting therapeutics. Chemical mapping data on its own does not allow to determine RNA structure, but Atomic’s foundation model ATOM-1 can learn some broad folding patterns from this large scale and diverse data, and when fine-tuned with a small number of 3D RNA structures, it provides a big boost in RNA-structure prediction accuracy [72].
A-Alpha Bio is a spinoff company that emerged from David Baker’s Institute for Protein Design at the University of Washington. The company has developed a new experimental cell-based platform that is vastly multiplexed and allows collecting millions of protein interaction affinities as training data for ML algorithms. Such large-scale protein interaction data is currently being used by A-Alpha and Dreamfold (a Canadian company founded in 2022 as a Mila spinoff) to develop generative ML algorithms for therapeutic protein design. Since affinity does not directly contain structural information, this type of data in combination with the task can be considered “black-box.”
Enveda (2019), a startup founded by Excursionautes (a term lovingly used for ex-Recursion employees that went on to found other startups, of which there are quite a few [73] and most of which are either Generation 2 or Generation 3) focuses on natural products derived from heterogeneous samples (such as plant extracts containing complex mixtures of small molecules) characterised via mass spectrometry (MS). In our terminology, MS spectra are to be considered “black-box data” that does not provide directly the molecular structures they contain. To cope with this very challenging task, Enveda applies AI methods to predict properties and select analog compounds directly from the spectra (i.e., in “black-box space”), allowing them to massively increase screening throughput [74]
VantAI (founded in 2019) is another Generation 3 company developing novel assays for AI-purpose systematic data generation. VantAI’s claim to fame is structural proteomics-based experimental technology allowing to produce structural interface data up to six orders of magnitude cheaper than X-ray or cryo-EM (reducing the cost of a protein-interface structure from currently up to 1M$ down to roughly the cost of an espresso), making the benefit of using black-box vs white-box data highly enticing. Such data on its own is “too sparse” to allow for the direct inference of structure (and thus similar to shotgun reads is considered black-box [64]), but it is its use in conjunction with specialised generative ML algorithms making it possible to produce up to 100–1000× larger and more diverse structural interface dataset than all of scientific history combined.
VantAI uses this technology for one of the most exciting but notoriously difficult-to-design drug modalities, proximity modulators (e.g., “molecular glues”) that induce, block, stabilise, or destabilise existing interactions by mimicking naturally occurring interfaces. Since natural proteins interact specifically with other proteins, it is very likely that there exists natural interaction produced by evolution we can imitate and learn from. As a result, besides a novel high-throughput data source the problem also has the beneficial characteristics of “degenerate” solution space and “distributional hypothesis” [75–76].
“Encheiresin naturæ nennt’s die Chemie,
Spottet ihrer selbst und weiß nicht wie.”
— Goethe, “Faust”
Alchemists used to believe in encheiresis naturæ (the “hand of Nature”), a semi-magical force that supposedly breathed life into living things [77]. We fortunately now have a much better understanding of biology eschewing the need to invoke magic. In some cases, we were able to trace the interactions between molecules to the effects on cells, leading to multiple successful drugs against diseases that just a few decades ago would spell a death sentence for the patient. However, we still largely lack a systematic way of connecting different biological scales, especially translating the knowledge of the low-level molecular processes to the functioning of cells, tissues, and entire organisms. Overly optimistically entrusting AI with somehow solving these problems is in a sense a return of alchemy, this time as “encheiresis machinæ” (“hand of the Machine”).
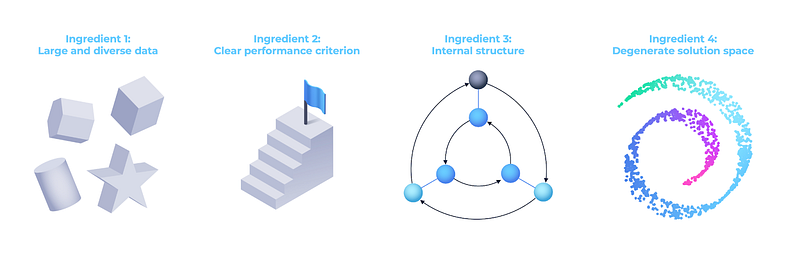
Artificial Intelligence on its own might be of only limited impact in biological sciences if applied blindly to problems without additional prerequisites. In this article we identify these prerequisites as the availability of large and diverse datasets; a clearly defined performance criterion; useful internal structure in the data (such as the distributional hypothesis) to enable unsupervised learning; and a degenerate solution space. Even within the biological problems better amenable to ML techniques, it is important to realise that any single “ImageNet-,” ”AlphaFold-,” or ”ChatGPT-moment” will not transform the pharmaceutical industry overnight: drug development is a long and arduous process, and even remarkable breakthroughs in “holy grail” biological problems such as predicting a protein structure are a far cry from all what is needed to delivering a successful drug to the market, and there remain many such “Grand Challenges” yet to be solved [78].
The Human Genome Project again provides worthwhile historical learnings here: it, too, solved one of the “holy grail” biological problems and triggered, at least in some, unrealistic expectations of being able to exploit genomic data to create a torrent of novel medicines that would cure many of the most complex diseases in one fell swoop. History shows that this did not happen [79]. Here, too, one big reason for the limited scope of success has to do with the fact that there is a lot of biology happening between a genetic variant being present in a genome and a disease phenotype and there remain many “Grand Challenges” yet to be solved in between. Similarly to AlphaFold, the HGP’s impact was not driven by answering any specific biological question, but as an enabling technology that allows to investigate “continuously arising new questions from the same data-rich source” [80].
We furthermore see the Human Genome Project as a successful early example of the benefit of developing experimental technologies producing “black-box” data unintelligible by human scientists and making sense only in tandem with appropriate computational methods. The road to Biology 2.0, in our view, entails a paradigm shift from traditional expensive and often scarce “white-box” data (Generations 0–2) to novel low-cost high-throughput “black-box” data (Generation 3) specifically designed to work with “black-box” AI-based approaches.
“Black-box” is usually a pejorative term in science, a modern-day version of “ignoramus et ignoramibus.” However, it does not imply that AI has to forgo the scientific knowledge we have acquired so far in favour of purely data-driven techniques. Quite the contrary, in the past years we have seen a trend of combining mathematical equations specific to a particular domain with general-purpose, machine-learnable components trained on experimental data. As eloquently formulated by Mohammed AlQuraishi, distilling knowledge about basic natural phenomena in the form of conceptual primitives often allows to model previously intractable complex systems, overcome the limitations of scarce, incomplete, and noisy data, and offer better generalisation and some level of interpretability. In biological and physical sciences, such approaches were given the name “differentiable biology” [81] and “physics-inspired learning.” Geometry offers a powerful framework to formalise some of these primitives through the language of invariance and symmetry — concepts underpinning Geometric Deep Learning and widely used in ML for chemistry, physics, and biology in the form of equivariant architectures. Again, AlphaFold2 is perhaps the most famous successful example of these principles.
While nimble startups will make significant progress towards transforming some of the steps of the long and arduous road to develop drugs, other “Generation 3” datasets will be less easy for them to obtain. As black-box data is still collected from the original sample under question (e.g., human activity data and other biomarkers to predict an individual’s phenotype) rather than a simplified model thereof (e.g., highly cultured cell lines, with questionable translational value), in some cases obtaining such data, although “black-box,” will remain inherently expensive. In particular, questions around complex human disease biology, which informs one of the most costly and important challenges — which disease target to go after [82] — would likely need to be collected at the human population scale. Coordinated governmental, academic, and industrial consortia, such as the NIH’s All Of Us Research Program or the UK Biobank will be required to achieve this and might take longer to emerge due to a potential aversion towards the “black-box” nature of Generation 3 data.
Nonetheless, despite the limited scope of each single of those Generation 3 breakthroughs, we find it exciting to see emerging patterns that companies can emulate and funding bodies and investors can use to effectively allocate resources. We believe any such company has the chance to make a significant dent and enable the discovery of highly impactful medicines — and, step by step, transform drug discovery as a whole.
[1] ChatGPT announcement by OpenAI, 30 November 2022.
[2] For example, Goldman Sachs predicted in March 2023 that generative AI technologies such as ChatGPT could replace 300M jobs.
[3] One of the most common drawbacks of ChatGPT is its tendency to “hallucinate” answers with high confidence (sometimes entirely making up sources), which is unacceptable in the academic realm and has already led to funny incidents.
[4] Sam Altman’s Economist interview, January 2024.
[5] Demis Hassabis’ talk at the Royal Society, April 2023.
[6] AlphaFold2 appeared in J. Jumper et al., Highly accurate protein structure prediction with AlphaFold, Nature 596:583–589, 2021 was published concurrently with a similar architecture RosettaFold, M. Baek et al., Accurate prediction of protein structures and interactions using a three-track neural network, Science 373:871–876, 2021. Both papers were featured on the covers of the respective journals.
[7] Such as structural clustering the entirety of the known protein universe to discover novel folds, see I. Barrio-Hernandez et al., Clustering predicted structures at the scale of the known protein universe (2023), Nature 622:637–645.
[8] D. Lowe, Docking with AlphaFold Structures: Oops, In the Pipeline, Science (2023).
[9] We give a few extreme examples of molecular dynamics (MD): the longest published result of an MD simulation is 1msec, performed on Anton, a massively parallel supercomputer designed by DE Shaw Research, see K. Lindorff-Larsen et al., How fast-folding proteins fold (2011), Science 334(6055):517–520. One of the largest protein systems simulated for 500μsec was a villin protein containing 20K atoms, using 200K distributed personal computer CPUs in the citizen science project Folding@home. Such simulations require extreme computational resources and are still beyond reach for real-world systems.
[10] P. W. Anderson, More is different (1972), Science 177(4047):393–396 beautifully wrote about emergent phenomena in physics: “The ability to reduce everything to simple fundamental laws does not imply the ability to start from those laws and reconstruct the universe […] The behavior of large and complex aggregates of elementary particles, it turns out, is not to be understood in terms of a simple extrapolation of the properties of a few particles. Instead, at each level of complexity entirely new properties appear, and the understanding of the new behaviors requires research which I think is as fundamental in its nature as any other”
[11] The first to publicly and controversially challenge the scientific methods in the age of “Big Data” was, to our knowledge, C. Anderson, The End of Theory: The Data Deluge Makes the Scientific Method Obsolete, Wired 2008. This idea of “post-theory science” was reiterated by Laura Spinney in The Guardian in 2018 following the AlphaFold breakthrough.
[12] AI-based science can still satisfy the falsifiability criterion of Popper. For a deeper discussion about the implications of AI in biology, see E. Lawrence et al., Understanding biology in the age of artificial intelligence (2024), arXiv:2403.04106.
[13] The “AlexNet” for image classification by A. Krizhevsky, I. Sutskever, and G. E. Hinton, ImageNet classification with deep convolutional neural networks (2012), NIPS. There were several contemporaneous efforts, notably by the group of Jürgen Schmidhuber.
[14] ImageNet Large Scale Visual Recognition Challenge was established in 2009 as an annual challenge consisting of the classification of millions of human-labeled images into 1000 different categories.
[15] The first Convolutional Neural Network (CNN) architecture was described by Y. LeCun et al., Backpropagation applied to handwritten zip code recognition (1989) Neural Computation 1(4):541–551, and was based on a previous work of K. Fukushima, Neocognitron: A self-organizing neural network model for a mechanism of pattern recognition unaffected by shift in position (1980), Biological Cybernetics 36:196–202, which in its own turn drew inspiration from the work of the Nobel Prize laureates D. H. Hubel and T. N. Wiesel, Receptive fields of single neurones in the cat’s striate cortex (1959), J. Physiology 148(3):574. The architecture in LeCun’s 1989 paper was unnamed; the term “CNN” appeared in a later paper, Y. LeCun et al., Gradient-based learning applied to document recognition (1998), Proc. IEEE 86(11): 2278–2324, where a 5-layer model called LeNet-5 was trained on 60K MNIST digit images. For comparison, Krizhevsky’s 2012 AlexNet was an 11-layer CNN trained on 1.2M images from ImageNet. Additional important changes compared to LeNet-5 were the use of ReLU activation (instead of tanh), maximum pooling, dropout regularisation, and data augmentation. See our previous blog post.
[16] First deep learning breakthroughs in natural language and speech used the Long Short-Term Memory (LSTM) architectures, introduced by S. Hochreiter. Untersuchungen zu dynamischen neuronalen Netzen (1991), Diploma thesis, TUM, and S. Hochreiter, J. Schmidhuber, Long Short-Term Memory (1997) Neural Computation 9(8):1735–1780. More recently, LSTMs were replaced by Transformers.
[17] A. Wissner-Gross, Datasets over Algorithms, 2016.
[18] OpenAI’s GPT-3 (released in 2020) had 175B parameters and was trained on 300B text tokens. The exact numbers for the most recent GPT-4 are not known, but rumors from reasonably trustworthy sources suggest a total of roughly 1.7T parameters and a gigantic training set of around 20T tokens.
[19] Although the datasets used to train AlphaFold2 were established long time ago, it is fair to say that they have grown significantly in recent years. The structural data used by AlphaFold2 came from the Protein Database (PDB), which was established in 1976 and first contained only 13 protein structures. As of 2024, the dataset contains slightly over 200K structures. The sequence data came from Uniprot (established in 2003 as a merger of multiple similar efforts), which has experienced an order of magnitude growth over the last decade (from around 25M protein sequences in 2012 to 250M as of 2024). AlphaFold2 was trained on 140K protein structures from PDB and 350K aminoacid sequences from UniRef, a subset of Uniprot. The model had 21M parameters and required 11 days of training on 128 TPUs. For comparison, Meta’s ESMFold 2 used an LLM pre-trained on 60M sequences; the largest model had 15B parameters and required 60 days of training on 512 NVIDIA V100 GPUs.
[20] A key architectural block of AlphaFold2 is the “invariant attention,” which builds upon the Transformer introduced by A. Vaswani et al., Attention is all you need (2017), NIPS, and the ideas of symmetry-aware architectures underlying geometric deep learning that were popularise around the same time e.g. by M. M. Bronstein et al., Geometric Deep Learning: Going beyond Euclidean data (2017), Signal Processing Magazine 34(4):18–42.
[21] As an important evidence of “algorithm over data,” it is instructive to compare the two versions of AlphaFold. The OpenFold team showed that when trained on ~1.5K structures, AlphaFold2 achieves better accuracy than AlphaFold1 on ~150K structures, thus in a sense being 100× more data efficient, see G. Ahdritz et al., OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization (2022), bioRxiv:2022.11.20.517210.
[22] In his 1972 Nobel lecture titled “Studies of the principles that govern the folding of protein chains”, Anfinsen stated that for proteins, “the native conformation is determined by the totality of interatomic interactions and hence by the amino acid sequence, in a given environment.”
[23] The stunning initial impact of AlphaFold in 2018 is perhaps best captured in Mohammed AlQuraishi’s blog post.
[24] Z. Lin et al., Evolutionary-scale prediction of atomic-level protein structure with a language model (2023), Science 379(6637):1123–1130.
[25] J. P. Roney and S. Ovchinnikov, State-of-the-Art Estimation of Protein Model Accuracy Using AlphaFold (2022). Phys Rev Lett. 129:238101.
[26] H.C.T. Nguyen et al., Benchmarking integration of single-cell differential expression (2023). Nature Comm. 14, 1570.
[27] K. Z. Kedzierska et al., Assessing the limits of zero-shot foundation models in single-cell biology (2023), bioRxiv 2023.10.16.561085.
[28] G. A. Landrum, S. Riniker. Combining IC50 or Ki Values from Different Sources Is a Source of Significant Noise (2024), Journal of Chemical Information and Modeling
[29] Note that technically, AlphaFold2 trains on “crops” of PDB and folding a single protein is roughly equivalent to folding a protein chain. The PDB roughly contains an order of magnitude more chains than PDBs, so one could consider the dataset to be more around ~1M chains, which is still tiny by ML standards, and it is also highly redundant.
[30] This estimate first appeared in C. Levinthal, How to Fold Graciously (1969) Mossbauer Spectroscopy in Biological Systems (for comparison, the number of protons in the observable universe is believed to be only around 1⁰⁸⁰). The fact that many natural proteins fold reliably and quickly to their native state despite the astronomical number of possible configurations is called the Levinthal’s Paradox.
[31] The “manifold hypothesis” posits that real-world high-dimensional data often lie along a low-dimensional latent manifold embedded in that high-dimensional space, allowing to describe the data by a small number of degrees of freedom corresponding to the intrinsic coordinates on the manifold. It is the central premise of nonlinear dimensionality reduction methods, see, e.g. J. B. Tennenbaum et al., A Global Geometric Framework for Nonlinear Dimensionality Reduction (2020), Science 290(5500):2319–2323.
[32] Gene diversity is primarily created through two processes during genetic recombination: independent assortment, where chromosomes from parents are intermingled in the offspring, and cross-over, where parts of chromosomes from parents are exchanged, potentially leading to gene duplications (or deletions) in non-homologous crossover. Additionally, there are point mutations. For biological optimisation to come up with a novel solution to a problem, the path that requires the least amount of generations is where large chunks of existing proteins can be copy-pasted, and small details (such as in the substrate-binding domain of an enzyme) can be mutated to achieve new effects. Arriving at such a solution “from scratch” by consecutive, independent random point mutations vs domain duplications, in turn, would require an exponential amount of additional iterations, and thus we see it happen more rarely in nature. For example, to arrive at a functional new enzymatic domain of size 200 amino acids, a single gene duplication event with 3–4 mutations might suffice, getting there in 1–2 replications, while arriving there from scratch would potentially require 200 consecutive exactly right mutations.
[33] S. Chen et al., Protein folds vs. protein folding: Differing questions, different challenges (2022), PNAS 120(1), e2214423119.
[34] This is by no means a new observation, and protein designers often operate with structural primitives rather than individual amino acids. For example, C. Mackenzie, J. Zhou, and G. Grigoryan, Tertiary alphabet for the observable protein structural universe (2016) PNAS 113(47): 7438–7447 found that most PDB protein structures can be captured by a few hundred of standard reusable building blocks (compact backbone fragment referred to as “tertiary structural motifs” or “TERMs”), pointing to a significant degeneracy of protein structures. Gevorg Grigoryan is now the CTO of a unicorn protein drug design company Generate Biomedicines.
[35] GPT-4 has been famously accused of doing just that: regurgitating and slightly modifying compressed information rather than generalising.
[36] Mohammed AlQuraishi argues that AlphaFold2 is an MSA-to-structure rather than a sequence-to-structure map. The former is a much easier problem than the latter, and it is likely that enough of the structure is embedded in the MSA to be just “read off” of it, see e.g., R. Rao et al., Transformer protein language models are unsupervised structure learners (2020), bioRxiv:2020.12.15.422761. MSA-to-structure was solved for some protein domains using unsupervised methods with very deep (100K–1M sequences) MSAs, see e.g. R. Sheridan et al., EVfold.org: Evolutionary Couplings and Protein 3D Structure Prediction (2015), bioRxiv:10.1101/021022.
[37] AlphaFold-latest accuracy drops 50% on fairly lenient “generalisation” cutoffs, see Fig 2a vs Fig 5a last column (“protein novel & ligand novel”) in the AlphaFold-latest technical report. These results are with 40% sequence identity and 0.5 Tanimoto similarity which can still include significant leakage — usual cutoffs are 30% sequence similarity (vs identity) and <0.25 Tanimoto similarity. While AlphaFold2-multimer presents a step change in accuracy for protein interface prediction, protein-ligand prediction cannot be considered nearly as “solved” as monomer structures.
[38] G. Corso et al., The Discovery of Binding Modes Requires Rethinking Docking Generalization (2023), NeurIPS MLSB Workshop.
[39] An exception might be natural products, however, data around these is even more sparse. For most proteins, there are no naturally evolved small molecules engaging them that we can “copy-paste.”
[40] W. Ahmad et al., ChemBERTa-2: Towards Chemical Foundation Models (2022) arXiv:2209.01712.
[41] Z. Harris, Distributional structure (1954) Word 10(23):146–162.
[42] For example, “the bison grazed at the … of the river”, makes it very clear that whatever is missing has to denote a location around a waterway, which allows one to learn the semantic meanings of the word “bank”, and also learn how to complete solutions by the most likely meaningful words or phrases.
[43] Take for example, a receptor protein on the cell surface. Most receptor proteins have the same setup of basic components: a transmembrane domain that anchors the protein in the cell membrane, an extracellular ligand binding domain that recognises its cognate ligand, and an intracellular signalling domain. Each of these components is then often composed of highly conserved elements that are found in that component across a wide variety of different proteins. For example, many intracellular signalling domains of such a receptor will contain a highly conserved kinase domain that is mostly identical in structure (and often sequence) since many of these receptors will have evolved through gene duplication events. Similar to our previous example with the river bank, a masked language modelling (or next token prediction) objective could look like the following simplified example: “[extracellular-domain]-[transmembrane domain]-[…]” . Due to the “degeneracy” of the solution space, the model will be able to learn both that “intracellular kinase domain” is highly likely to go into the […], and learn the functional (semantic) relevance of a kinase domain. The same goes for specific residues within a domain — for its catalytic function, the kinase domain will need to contain a characteristic set of amino acids (e.g. the “DFG” loop, comprised of the Asp-Phe-Gly (DFG) motif) to bind its substrates, the semantic meaning of which can be learned through token masking due to their conserved appearance within these abundant domains.
[44] J. W. Schafer et al., Sequence clustering confounds AlphaFold2 (2024), bioRxiv:2024.01.05.574434.
[45] D. Chakravarty et al., AlphaFold2 has more to learn about protein energy landscapes (2023), bioRxiv:2023.12.12.571380.
[46] AlphaFold2 used a special structure loss, the Frame Aligned Point Error (FAPE), which is roughly obtained by averaging the prediction error at every atom viewed under a number of different frames.
[47] Zhang et al., Protein language models learn evolutionary statistics of interacting sequence motifs (2024), bioRxiv:2024.01.30.577970v1. It supports the idea that AlphaFold and ESMFold approach protein folding via similar retrieval mechanisms. ESMFold, which replaces MSA used in AlphaFold with large-scale pre-trained single-sequence LLMs, seems to directly learn equivalent information to the direct coupling and covariance information obtained from MSA through the LLM pre-training (although MSA-based methods are still more accurate). The paper explores the nature of the retrieval process further, arguing that retrieval works by learning evolutionary statistics of short (20–40 residue) sequence motif pairings, shedding some light on how the posited retrieval might work.
[48] R. Wu et al., High-resolution de novo structure prediction from primary sequence (2022) bioRxiv:2022.07.21.500999.
[49] To the best of our knowledge, the term was first introduced by G. Carr, Biology 2.0 (2011), The Economist.
[50] See Nvdia blog, 12 January 2024.
[51] We should note that the distinction between “generations” (especially between Generation 2 and 3) is continuous rather than discrete. Furthermore, companies have evolved over time and some of those that started as Generation 2 have at least in part moved to Generation 3 methods.
[52] A. Karpathy, Software 2.0 (2017), Medium blog post.
[53] C. Horan, NLP Datasets: How good is your deep learning model? (2020), Medium blog post
[54] Optimising kinase inhibitors, while certainly helpful, is where medicinal chemists have the least struggle. A very nice summary of this point is given by M. Volkov et al., On the Frustration to Predict Binding Affinities from Protein-Ligand Structures with Deep Neural Networks (2022) J. Med. Chem 65(11):7946–7958.
[55] See, e.g., the following quote from Derek Lowe in [8]: “small molecules have a far greater variety of structural motifs and a greater variety of interaction modes with the proteins they’re binding do. You don’t have to worry about halogen bonds when you’re doing protein structure, just to pick an obvious example, because no natural proteins have chlorines or bromines in them. Similarly, there are plenty of other things that rarely or never come up: torsional angles of sulfonamide linkages, pi-interactions of pyridines, the polarity of sulfoxides, the conformational flexibility of azepane rings… we’re an imaginative bunch, we organic chemists, and a couple of billion years of natural product evolution have provided further structures that keep surprising even us. So I think that there are a lot more structural situations to work though, while at the same time there are nowhere near as many data points as you have for straight protein structure. The number of small-ligand-bound structures in the PDB is only a small fraction of the total number of PDB structures — you’d want it to be several times the size to have a better shot at AlphaFolding your way to the solutions, in my view.”
[56] See e.g. the blog post by M. Strasser The Business of Extracting Knowledge from Academic Publications (2021).
[57] See e.g. the blog post by S. Bannon, Defensibility in the Age of AI (2023).
[58] A more recent evolution of Generation 2, which we call “Generation 2+,” also tries to replace the end human consumer with a machine. Various “lab-in-the-loop” approaches fall into this category.
[59] M.-A. Bray et al., Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes (2016), Nature Protocols 11:1757–1774.
[60] K. McCloskey et al., Machine Learning on DNA-Encoded Libraries: A New Paradigm for Hit Finding (2020), J. Med. Chem. 63(16):8857–8866.
[61] In its early days, cryo-EM was given the derisive nickname “blobology”, due to its poor resolution. A breakthrough came with the emergence of better electron detectors, leading to a revolutionary impact in structural biology and the 2017 Nobel Prize.
[62] E.g. generative diffusion models introduced by J. Sohl-Dickstein et al., Deep Unsupervised Learning using Nonequilibrium Thermodynamics (2015), ICML and flow matching by Y. Lipman et al., Flow matching for generative modeling (2023) ICLR.
[63] A classical example is Kepler’s law of motion. Johannes Kepler studied a trove of astronomical observations produced by the astronomer Tycho Brahe, finding that the motion of Mars contradicted the Copernican theory of circular orbits of planets. He hypothesised that the orbits are elliptical, which he then confirmed by further observations.
[64] Since we have not encountered the term “black-box data” before, we need to make it more precise. We consider the data together with the function (or task) for which it is acquired. We say that data is “black-box” if it does not directly contain sufficient information for a human about the function it is collected for. “Black-box” data is not always necessarily “gibberish” to humans — it just means that humans cannot go directly from the data to evaluating the hypothesis, but must entirely rely on a “black-box” ML model to link data and hypothesis. In our example of shotgun sequencing [69], the data (small DNA fragments) have a clear meaning to a human (a short sequence of nucleotides), however, a human cannot use them on their own to get a good idea about the task (full genome reconstruction) — this is only possible with additional computation that is typically beyond human ability (a sequence alignment algorithm such as BLAST). Similarly, in an event camera [67], the data (time and position of a pixel increasing or decreasing its brightness) does not provide much information to a human about the task (image or video reconstruction). Considered from the viewpoint of data/function, even exactly the same data source can be “black-” or “white-box” in different contexts: for example, a cryo-EM structure is white-box for the task of structure prediction or docking, but likely to be black-box for predicting higher-level cellular functions. This, in our view, constitutes a key criterion to make a somewhat elusive distinction between Generation 2 and 3 companies: it is not whether the data source is designed to be used by ML (a characteristic of Generation 2, e.g., in the words of Insitro CEO Daphne Koller: “To enable the machine learning, we […] invest heavily in the creation of our own datasets using high throughput experimental approaches, datasets that are designed explicitly with machine learning in mind from the very start”) but whether, even hypothetically, the data source could be used by a human to carry out the downstream task. For example, it is conceivable that a human scientist could, in principle, make predictions by studying cell image data (though it is ML that makes this task feasible in practice). On the other hand, random projection data used in compressed sensing [68] will be absolutely uninformative for a human. In reality, this is not a sharp distinction, and many companies that started as Generation 2 (e.g. Recursion [71]) are likely to gradually shift towards Generation 3 as their experimental and ML technology evolves.
[65] As an example how ML-focused data tradeoffs can unlock new experimental setups, consider protein docking problems, which are often seen as “NP-hard,” in the sense that it is hard to find a solution but easy to verify it [38]. Bringing the solution space from an intractable or possibly infinite number of conformations to a few thousand or hundred will not help a human, since humans will still not be able to explore this solution space, but will already give most information required to solve the problem for a ML model which can search through the solutions and verify them easily. Only needing to reduce solution space, rather than directly measuring the solution experimentally, in turn, allows to drastically reduce the required resolution of the assay to expand the scale. A practical example is using AlphaFold2 predicted structures in cryo-EM refinement, improving the quality of lower resolution cryo-EM maps with the help of ML in order to unlock the possibility of determining much larger complexes than previously possible at high accuracy, see e.g., P. Fontana et al., Structure of cytoplasmic ring of nuclear pore complex by integrative cryo-EM and AlphaFold (2022) Science 376.
[66] The use of the original biological system rather than a simplified one is what distinguishes “Generation 3” from approaches relying on large-scale simulations allegedly adopted by Isomorphic Labs. We do believe that simulated data can be very useful in some problems, and most likely, the winning approaches will rely on different types of synthetic and experimental data from multiple sources.
[67] Event cameras are special image sensors that respond to local changes in brightness. Each pixel stores a reference brightness level and continuously compares it to the current brightness level. If the difference in brightness exceeds a threshold, that pixel resets its reference level and generates an event: a discrete packet that contains the pixel position and timestamp.
[68] Compressed sensing is a class of signal reconstruction techniques based on sparsity, see e.g. E. J. Candès et al., Stable signal recovery from incomplete and inaccurate measurements (2006) Communications on Pure and Applied Mathematics 59(8):1207–1223.
[69] The Human Genome Project (HGP) was established in 1990 with the goal of sequencing the full human genome within the following 15 years at the cost of $3B. HGP used a systematic, hierarchical mapping and sequencing approach. A private company Celera founded by Craig Venter proposed a controversial and radically new whole-genome shotgun approach, allowing to lower the cost by an order of magnitude and finish the sequencing of the genome two years earlier. See J. Shreeve, The Genome War: How Craig Venter Tried to Capture the Code of Life and Save the World (2005) and C. Venter, A Life Decoded: My Genome: My Life (2009).
[70] In the past years, a new kind of research organisations (“Focused Research Organisation” or FRO) started emerging, combining the best features of non-profit academic institutions, big corporations, and startups. One such recent example is Future House, funded by Eric Schmidt.
[71] P. Lubroth, Scaling Biology 001: Chris Gibson, Co-Founder and CEO of Recursion Pharmaceuticals (2023), Decoding Bio.
[72] Boyd et al., ATOM-1: A Foundation Model for RNA Structure and Function Built on Chemical Mapping Data (2023) bioArXiv:2023.12.13.571579.
[73] Including companies like Noetik.AI, Enveda, and Leash.bio.
[74] G. Voronov et al., MS2Prop: A machine learning model that directly predicts chemical properties from mass spectrometry data for novel compounds (2022), bioRxiv:2022.10.09.511482.
[75] The space of protein interfaces is known to be degenerate, see e.g. M. Gao and J. Skolnik, Structural space of protein–protein interfaces is degenerate, close to complete, and highly connected (2010), PNAS 107(52):22517–22522, who attribute this degeneracy to the “packing of compact, hydrogen-bonded secondary structure elements”, which generates “relatively flat interacting surfaces whose geometries are highly degenerate.”
[76] M. Gao et al., Structural space of protein–protein interfaces is degenerate, close to complete, and highly connected (2010) PNAS 107(52): 22517–22522.
[77] The greco-latin term “ἐγχείρησις naturæ” (literally “handgrip of nature”) goes back to medieval alchemical treatises. Jacob Reibold Spielmann wrote in his Institutiones chemiæ (1763) thus: “Natura plures noverit encheireses substantias uniendi, quas nos sive ignoramus sive imitari non valemus” (”Nature knows many encheireses of uniting substances, which we either do not know or are unable to imitate”). The term “encheiresis naturæ” in this context should most accurately be translated as “natural process,” or what modern science would call a “chemical reaction.” Since these were still poorly understood in the eighteenth century, “encheireses” were endowed with spiritual or even magical properties. The poet and natural philosopher Johann Wolfgang von Goethe, who studied under Spielmann in Strasbourg during 1770, disliked this buzzword so much that he famously parodied it in Mephistopheles’ pseudo-scientific speech in Scene IV of the first part of Faust:
“Encheiresis naturæ, Chemistry calls it,
Mocking itself, not knowing what befalls it.”
(in our epigraph, the original German phrase “nennt es die Chemie” requires the accusative declension of “encheiresis,” for which Goethe uses the correct Greek one, “encheiresin” — apparently unbeknownst to many translators who copy-paste a grammatical form inexistent in English).

[78] D. Lowe, Why AlphaFold won’t revolutionise drug discovery (2022), Chemistry World.
[79] PCSK9, first identified in the year of the conclusion of the Human Genome Project (via a hyperfunction mutation in familial hypercholesterolaemia), is just one of genomics grand successes, making a big impact on cardiovascular care today. Furthermore, there is recent evidence that genomically-validated targets have higher success, see D. Ochoa et al., Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs (2022) Nat. Rev. Drug Discovery 21, 551.
[80] L. F. Abbott et al., Mind of a Mouse (2020) Cell 182(6):1372–1376.
[81] “Differentiable biology” is a term proposed by M. AlQuraishi and P. K. Sorger, Differentiable biology: using deep learning for biophysics-based and data-driven modelling of molecular mechanisms (2021), Nature Methods 18:1169–1180, by analogy to “differentiable programming,” a superset of deep learning architectures trainable by backpropagation rules. “Differentiable” here refers to functions with defined derivatives.
[82] Two memes from Keith Hornberger convey a somewhat sceptical view of drug hunters on the promise of biological AI to transform drug discovery and the crucial question of selecting the right disease targets:

Luca Naef is a founder and CTO of Vant AI. Michael Bronstein is the DeepMind Professor of AI at the University of Oxford, Chief Scientist at Vant AI, and also a scientific advisor to Dreamfold and Recursion. Opinions expressed in this article are exclusively of the authors and do not necessarily reflect those of the aforementioned companies. We are grateful to Mohammed AlQuraishi, Nathan Benaich, Dominique Beaini, Joey Bose, Alex Bronstein, Martin Buttenschoen, Zach Carpenter, Ian Churcher, Bruno Correia, Gevorg Grigoryan, Stephan Grill, David Healey, Matt McPartlon, Padma Kodukula, Maksym Korablyov, Patrick Schwab, Oliver Stegle, Nico Thomä, and Georg Winter for multiple proof-reading and editing iterations and providing insightful comments that have helped shape and sharpen the concept of this post. We also thank Jitka Hrabálková for the beautiful illustrations and Sitabhra Sinha for providing an English translation of “Faust” (based, in turn, on the translation into Russian by Boris Pasternak), which, among all the versions we are familiar with, captures most poetically the difficulty of bridging biological scales.





