
Revisiting the bioflocculant hypothesis: A new Alzheimer’s culprit
When I was a graduate student, the microbiology section of our course work was run by a microbiologist who studied the bacteria involved in the development of gum disease. As a graduate student doing my PhD in neuroscience and studying one of the most feared diseases known to humans- Alzheimer’s disease- I always found it odd that someone could devote their life to studying gum disease and the microbiome responsible for its progression. How underwhelming, I had thought. It seems, I am beginning to realize, that I was premature in writing off that particular area of biomedical research, especially now, in the slow, but ever evolving world of Alzheimer’s disease.
I will start out by saying that the field of Alzheimer’s disease has been desperately searching for its etiology for well over a century. The community has accepted the Amyloid Cascade Hypothesis as the only possible explanation for the disease’s pathogenesis. Unfortunately, this hypothesis has not delivered on providing a sufficient explanation for the onset of the disease nor a target against which therapeutic intervention can be implemented.
The reasons for the above failures are probably several fold. However, the persistence of the central dogma in the field, what some snarky researchers have dubbed “The Church of Holy Amyloid,” by those who worship it, has been the major stagnating force slowing progress. In brief, the hypothesis goes like this: overproduction or underclearance of the amyloid-beta protein results in its aggregation into either large plaques or small, soluble toxic forms. The creation of these plaques/soluble forms is then believed to cause destabilization of the microtubule associated protein tau (a structural component of neurons). This destabilization throws the internal physiology of neurons into disarray leading to their dysfunction. Unfortunately, the proof of this mechanism in humans is lacking, especially since shutting down these processes with drugs has completely failed.
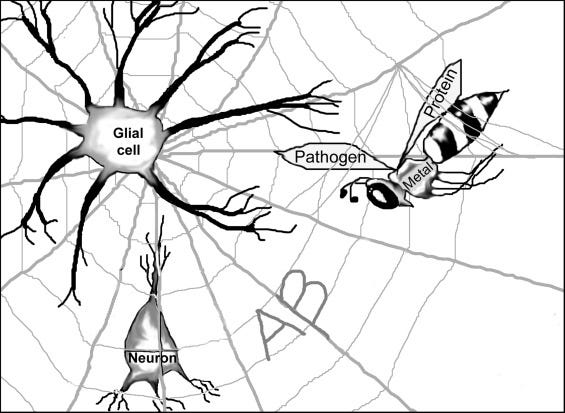
Fortunately, one often overlooked theory seems to be making a comeback, although the name by which it goes is not well known. In 2002, Stephen Robinson and Brenda Bishop proposed the bioflocculant hypothesis to explain the role of amyloid-beta in Alzheimer’s disease pathogenesis. It’s a simple, broad hypothesis with testable questions, as we are now witnessing. It posits that secreted amyloid-beta aggregates, acting like a spider web and ensnaring any foreign or domestic pathogenic material. This makes for an easy graphic to visualize (see figure above). Of the little nasties one would expect this web to trap, bacteria or other microbes are high on the list (as well as unwanted proteins, blood products, and metals). Once this pathogenic material is immobilized, microglial cells, the brain’s proverbial garbage men of the brain, can engulf and dispose of it.
The beauty of this thoery is really two-fold. The first, and most intriguing, is it gives a function to the amyloid-beta protein. This makes sense in light of evolution. It is likely that the mammalian brain, especially one as long-lived as a human’s, evolved a system to protect itself from a lifetime of invading environmental detritus. Second, it can explain why the amyloid-beta protein aggregates and is found in the majority of elderly with and without cognitive decline. The evidence Robinson and Bishop present in their two papers is striking and there has been no shortage of evidence published since, corroborating their theory.
It is important to think critically on the two points above. Basically, together they indicate a high likelihood that the aggregation of amyloid-beta is a response to some pathological insult, rather than being the insult itself*. In other words, a the red herring. This has ultimately born out in the myriad of failed clinical trials attempting to remove amyloid-beta from the brain of Alzheimer’s patients. In fact, some patients ended up with worsening symptoms.
The most recent study forcing researchers to rethink how Alzheimer’s disease develops was published last week in Science Advances. The authors of the new study were able to show that a bacterium found in our mouths around our teeth may have virulence factors capable of inciting a cascade of events that could lead to Alzheimer’s disease. And it fits into the bioflocculant hypothesis. The bacterium is called P. gingivalis and is the bacterium that I so foolishly dismissed at the beginning of this story. To the point, this particular bacterium secretes toxins called gingipains. The researchers showed that not only are these toxins found in the cerebral spinal fluid (CSF) of Alzheimer patients, but that the level of gingipains in the brain measured at autopsy was moderately correlated with the diagnosis of Alzheimer’s disease. A correlation did not exist with other neurodegenerative diseases. The fact that the toxins (as well as the bacterial DNA) can be found elevated in Alzheimer CSF versus controls and other diseases makes a strong case for bio marker development.
After establishing that the toxins and the bacteria were found in both brain and CSF of human Alzheimer’s patients, they turned to a mouse model of the disease to determine if blocking these toxins could stop disease progression. Indeed, by inhibiting gingipains with small molecule inhibitors developed by the biotech company Cortexyme, the authors could block neurotoxcity both in cultured neurons and in mice. Interestingly, broad spectrum antibiotics had no affect. However, of particular interest was the affect amyloid-beta had on microbial levels. The levels of amyloid rose in concordance with the length of infection with P. gingivalis. When challenged with amyloid peptides in culture, the bacteria were susceptible to its anti-microbial properties.
These results fit nicely with the current thoughts on the anti-microbial properties of amyloid-beta. One can imagine then that the increased levels of amyloid found in Alzheimer patients as a marker of a failed response by amyloid-beta to suppress some environmental insult (such as P. gingivalis or human herpes virus infection). On the other hand, the thirty percent of cognitively normal adults with high amyloid levels could have a better functioning amyloid-beta peptide, or at least a more efficient response by the immune system to gobble up the inciting pathogen that gets entwined in the amyloid web. There are a number of studies showing the variance in immune system genes of Alzheimer patients versus normal aged controls, but that’s a different story for a different time.
Of interest here is the potential variability of certain people to fight off Alzheimer’s disease-causing insults with their personal flavor of amyloid. Could it be that the highly touted paper from 2012 that found a protective mutation in the amyloid precursor protein, was, instead of proving amyloid as the culprit, actually proving the adaptive response capabilities of an evolved, anti-Alzheimer’s disease peptide? I think so. For now, though, it will be interesting to see how the small molecules used in this preclinical study will pan out in upcoming clinical trials. Cortexyme has already completed a Phase 1b trial and will be recruiting for a Phase 2/3 trial this year. Hopefully, they can change the tide of this stagnated field. Now, go see your dentist.
*This does not apply to the familial form of Alzheimer’s disease.





