Bioinformatics 1: K-mer Counting
A challenging yet intriguing interdisciplinary problem

K-mer counting is an interesting yet challenging problem in bioinformatics. In this article, we’ll talk about what k-mers are, the problem of k-mer counting, its applications, and some interesting insights from the computer science perspective.
What are k-mers?
In simple terms, k-mers are substrings of length k in a given string (can be DNA, RNA, protein, or any string sequence). Since our interest is towards bioinformatics, we will converge our attention to k-mers in a DNA sequence.
Consider the DNA sequence “ACGAGGTACGA” which consists of 11 nucleotides. Let’s try to obtain all the 4-mers (substrings of length 4) in this DNA sequence.
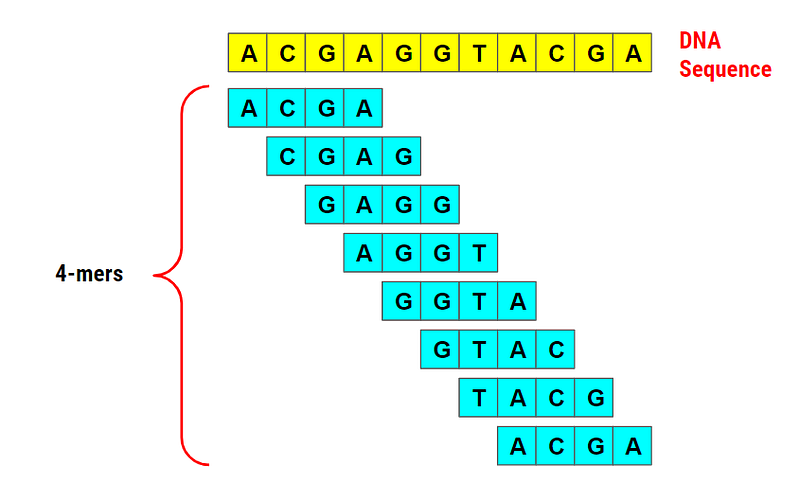
The idea is simple. We create a window of length 4 and slide it from left to right, shifting one character at a time. If the length of the given DNA sequence is N, we would end up with N - k+1 k-mers.
Total no. of k-mers = N - k + 1
In the above example, the given DNA sequence is 11 characters long (N=11) and k = 4, thus we get eight 4-mers (11 - 4 +1).
The Problem of k-mer Counting
K-mer counting is not one of those labyrinthine problems which are complicated and confusing. It is a very simple problem where you count the frequency of each k-mer in the given sequence. We usually look for three different types of frequencies: total count, distinct count, and unique count.
Let’s consider the aforementioned DNA sequence again.
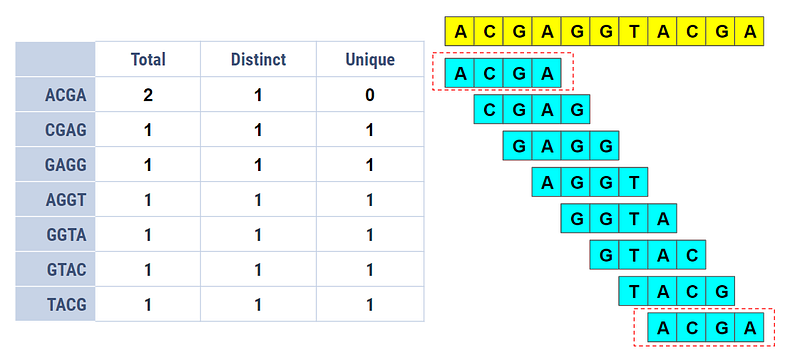
The Total Count is simply how many times each k-mer has appeared in the given sequence. Except for ACGA, which has appeared twice, the rest of the 4-mers have appeared only once.
The distinct k-mers are counted only once regardless of how many times they appear. For example, even though ACGA has appeared twice, we count it only once. The Distinct Count provides us the information of whether a k-mer has appeared or not (not how many times it has appeared).
The unique k-mers are those which appear only once. In the above example, since ACGA has appeared twice, its unique count is zero. The Unique Count reveals which k-mers have appeared only once.
The output of k-mer counting would usually be in tabular form as in the figure above or a histogram.
Applications of k-mer Counting
This section is worthy enough to write a separate article. At this point, I’ll list some of the applications where k-mer counting is used.
- Genome assembly
- Sequence alignment
- Sequence clustering
- Error correction of sequencing reads
- Genome size estimation
- Repeat identification
K-mer Counting from the Perspective of Computer Science
At a very high level, there are only three main steps in k-mer counting.
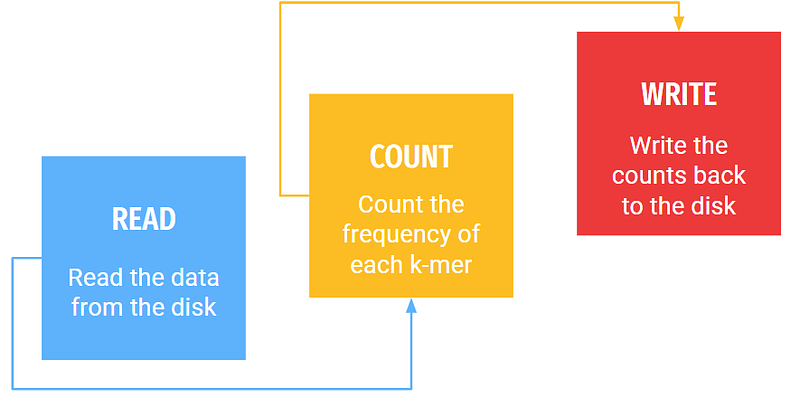
How difficult could it be!!!! Read the data, count it, and write the results back to the disk!!! What’s the need for all the fuss!!!
Well, let us do some math to figure it out.
We have k-mers and their corresponding counts. Suppose k=3

Since there are only 4 nucleotides {A, C, G, T}, we can represent each character using only 2 bits. Let’s assign 1byte / 8bits for the count.
Then 8+ 6 = 14 bits per row.
You would have 4³ rows in total.
Therefore, total memory requirement = 4³ (8+6) = 896 bits / 112bytes
What happens when you increase the k-value? The next graph would leave us in awe.
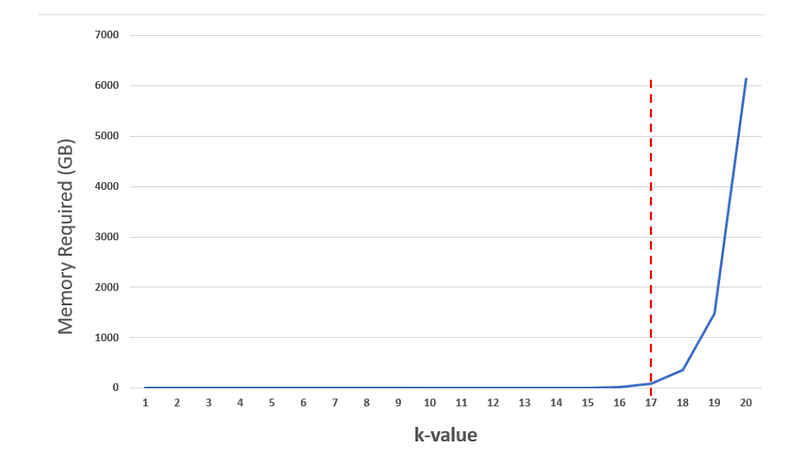
For k=20, you need almost 6000GB of RAM, which is massive!!! This memory bottleneck is what makes k-mer counting a very challenging yet intriguing for computer scientists.
I’ll leave the different approaches adopted to solve this problem to a different article.
I hope this article gives a very high level but a clear idea about k-mer counting!!!