
An Algorithm That Saved Biology and Shaped Modern Medicine
Finding patterns is an essential part of research in Biology.

Be it finding repeating patterns in DNA, RNA, or in a string of amino acids in a protein, the applications of pattern matching can be found in multiple places.
Let’s dive in deep!
The Human DNA
The genetic information of a cell is found in its nuclei (also in mitochondria, but that is for some other day).
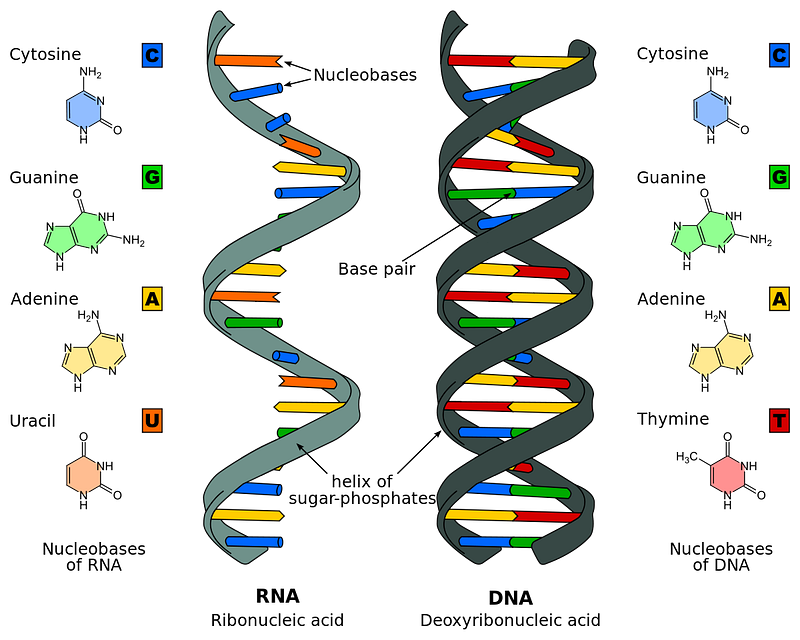
This information is in form of DNA (deoxyribonucleic acid) that is structured as chromosomes.

Human DNA is a polymer that is composed of two chains formed by monomers called nucleotides.
This polymer is very long and contains millions of nucleotides.
These chains twist around each other and form a double helical structure.
There are four types of Nucleotides in each chain and these are:
- Adenine (A)
- Thymine (T)
- Guanine (G)
- Cytosine (C )
A Ground Breaking Research
Let’s say that our hypothetical research group found out that a sequence of nucleotides (gene)ATTCCA , if present in one’s DNA causes early heart attacks.
It’s all over on the television and people are gathering to get their DNA tested.
If we sequence one’s DNA, we will get a long string of millions of nucleotides.
ATCGATGCAAGGTCGATCGATGCGCGCTAGCGATCGGCATCGATCGAGTCGATCGATCGATCGA...Searching for our sequence of interest is a time-taking and tedious task for a human being and hence we need a computer to help with this.
Programmer Stepping In
Let’s say that we have a programmer in our research group.
We ask them to write an algorithm that can find this pattern in our long string of nucleotides.
Problem Statement
A problem statement is formulated that explicitly mentions what we need our computer to do.
Given two strings:
- the string of all nucleotides in the DNA (
dna)
- the string of the gene of interest (
gene)
Find all occurrences of
geneindna
A Naïve Solution
The programmer starts with a brute force approach to solve this problem.
Steps
- We save the length of
dnaindna_lengthvariable and the length ofgeneingene_lengthvariable. - We set
pointer_1to the beginning ofdnastring and increment it using aforloop till indexdna_length — gene_length + 1 - We set
pointer_2to the beginning ofgenestring and increment it using awhileloop - The
ifstatement checks if nucleotides at both pointers’ indexes are equal. - If this is the case,
pointer_2is incremented, and if notpointer_1is incremented. - If
pointer_2transverses the whole length ofgeneand is able to match with the last nucleotide, we print that a match has been found.
Great job!
But is this algorithm efficient?
This algorithm runs in:
- Time complexity of O (
gene_length * (dna_length — gene_length + 1)) because we are looping through both of the strings. - Space complexity of O(1) because we are not creating any new intermediate data structure to be used by our algorithm.
An Efficient Approach: Knuth–Pratt-Morris algorithm
Steps in the Knuth-Pratt-Morris Algorithm!
As defined by Wikipedia, this algorithm searches for occurrences of a string within another string by employing the observation that when a mismatch occurs, the first string itself embodies sufficient information to determine where the next match could begin, thus bypassing re-examination of previously matched characters.
The time complexity of this search algorithm is O(dna_length + gene_length ).
The space complexity of this search algorithm is O(gene_length ).
But how does the algorithm do this in linear time?
The Magic Ingredient
To perform the pattern search in linear time, the algorithm performs a step where the gene string is pre-processed to construct an intermediate array that contains the longest proper prefix that is also a proper suffix of the string.
Confused? Let’s clear things up.
Example 1
Let’s take a gene string i.e. ATGA
The prefixes of this string are A , AT , ATG , ATGA .
The suffixes of this string are A,GA , TGA , ATGA .
When we consider the proper prefixes and suffixes, we exclude the whole string prefix and suffix, respectively.
Hence, the proper prefixes of this string are A , AT , ATG (excluded ATGA ).
The proper suffixes of this string are A,GA , TGA (excluded ATGA ).
Example 2
Let’s take another gene string i.e. ATATAGTA .
Let’s make an array that contains longest proper prefix that is also a proper suffix of the string.
|A|T|A|T|A|G|T|A|[0,0,1,2,3,0,0,1]Look at the substring till index 2 i.e. ATA
The proper prefixes of it are A , AT
The proper suffix of it are A , TA
The longest proper prefix that is also a proper suffix of this substring is A . The length of it is 1 . Hence, we put 1 at this index in the array.
Example 3
Look at the substring till index 4i.e. ATATA
The proper prefixes of it are A , AT , ATA , ATAT
The proper suffix of it are A , TA , ATA , TATA
The proper prefixes that are also proper suffixes of this substring areA and ATA.
The longest of them i.e. ATA has a length of 3 .
Hence, we put 3 at this index in the array.
Creating The Intermediate Array
Let’s write up a function that generates this intermediate array.
This function uses two pointers i and j at indices 1 and 0 respectively.
This function will return the intermediate array i.e. [0,1,2,0,1,2,3,3] .
Searching Our Gene of Interest in the DNA
We will use two pointers for this.
i is a pointer on the dna string.
j is a pointer on the gene string.
While searching we try to match each nucleotide in the gene with the nucleotide in the DNA.
But this will make us loop through both strings like in the naïve approach.
To avoid this we use our intermediate array to decide the next characters to be matched (avoiding characters that will match anyways) i.e. by changing j according to our generated intermediate array arr .
Let’s try it out!
This outputs to:
Gene found at index 0
Gene found at index 2
Gene found at index 4
Gene found at index 6
Gene found at index 8
Gene found at index 10
Gene found at index 12
Gene found at index 14And this is how Knuth-Pratt-Morris algorithm makes things easy and saves time for biologists!
Resources
Thanks for reading this article!
If you are someone new to Python or programming overall, check out my new book called ‘The No Bulls**t Guide To Learning Python’ below:





