
5 Cool AI-Powered Drug Discovery Tools
Harnessing the power of machine learning with Open Source tools for drug discovery
Proteins, made up of chains of amino acids, are the building blocks of life, drug discovery tasks generally include predicting protein folding, docking, and interactions. This has several applications from vaccines to other kinds of drugs especially for neurodegenerative diseases, and material science. As in other fields, biotech, biology, and medicine have been positively affected by the advancements in machine learning and quantum computing. The main advantages of in-silico drug design are related to the infinite variations which can be simulated and tried compared to tenfold costs and time in trial-and-error tests in the real world. Optimization procedures based on optimization look like the one shown in the video below:
Among the most popular tool for this kind of optimization is Ab-initio Rosetta which is based on the Montecarlo method. More recently quantum annealers are outperforming this kind of optimization by taking advantage of processes like quantum tunneling. Further applications based on machine learning techniques exist. I will skip commercial tools as Exscientia and quantum machine learning. If you are interested in this read this article below
Instead, in this article I list the most well-known open-source tools for drug discovery, linking their Github repositories and official papers:
- AlphaFold
- Generative Tensorial Reinforcement Learning (GENTRL)
- DeepChem
- Open Drug Discovery Toolkit (ODDT)
- Unified rational protein engineering with sequence-based deep representation learning (UNIREP)
AlphaFold
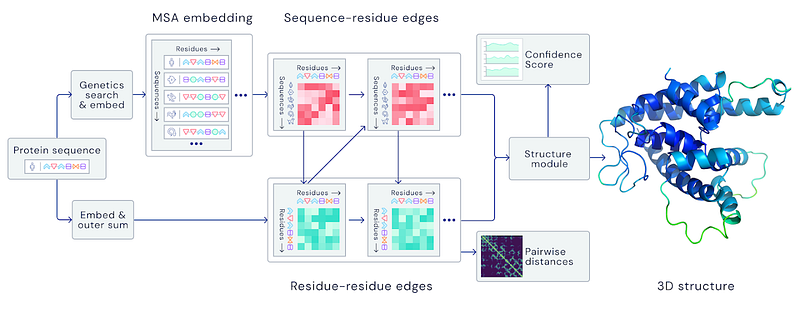
Biotech News in the latest 2 years has been focused on this tool from Deepmind. Indeed the two software versions from DeepMind’s program outperformed the other 97 teams in a biennial protein-structure prediction challenge Critical Assessment of Structure Prediction (CASP) in the 13th and 14th editions. AlphaFold is practically a neural network that predicts the structure of a protein’s spatial graph given its DNA. It is used as input genetic sequences and with the combination of multiple sequence alignment, geometric representations, and deep learning to obtain the protein’s structural graph. The 14th CASP edition was focused on the ORF3a protein of the SARS-COV2 protein but tested also in other related COVID19 proteins.
As tons of reviews about this tool have been already all over the media, I limit to share the repository for the first version here which won the CASP13 in 2018 which is based on Tensorflow 1.14. The original paper was published in Nature 2020.
Alphafold 2 has been preseted but a “proper article” and the official repository has not been published. Until there will be, the official publication is the abstract of Jumper et al. at CASP14 and the unofficial implementations based on Pytorch available at https://github.com/lucidrains/alphafold2
The major difference between the tools is that AlphaFold 1 and AlphaFold 2 are in the used neural network architecture. Version 1 used concurrent neural networks (CNNs) and version 2 uses Transformers.
Generative Tensorial Reinforcement Learning
The generative Tensorial Reinforcement Learning (GENTRL) tool is developed by Insilico Medicine to accelerates the process of experimental validation. At the core, there is a variational autoencoder. The framework uses tensor decompositions to encode the relations between molecular structures and their properties and to learn on data with missing values. As the name said it is based on reinforcement learning and a typical pipeline as below.
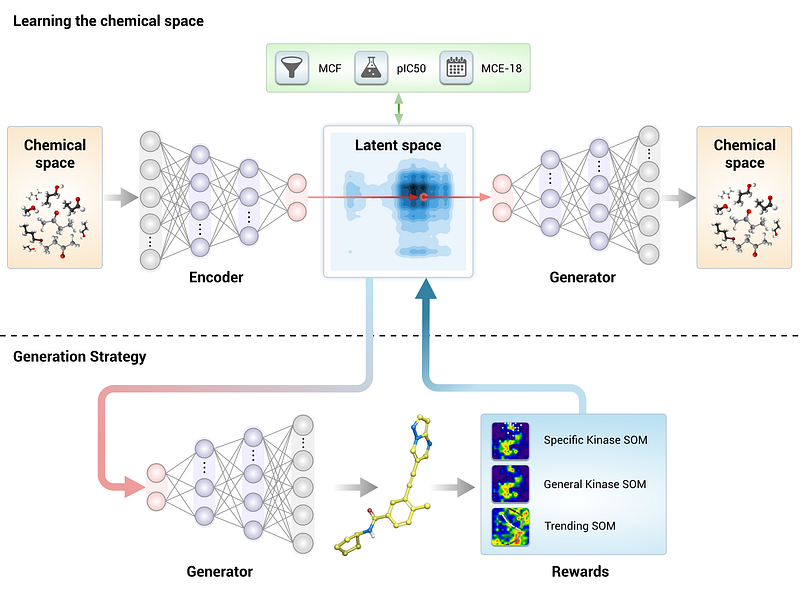
The tools are available on the repository, and it is based on Pytorch and RDKIT. It has been used to discover potent inhibitors of discoidin domain receptor 1 (DDR1), a kinase target implicated in fibrosis and other diseases. In the reported experiment, the entire process took only 21 days with the then-available computers.
DeepChem
DeepChem is an open-source deep learning framework aiming at democratizing drug discovery. The features listed on their website are
Predict the solubility of small drug-like molecules
Predict binding affinity for small molecule to protein targets
Predict physical properties of simple materials
Analyze protein structures and extract useful descriptors
Count the number of cells in a microscopy image
It uses both Google TensorFlow and scikit-learn machine learning tasks and RDKit for operations on molecular data, like converting SMILES strings into molecular graphs.
Open Drug Discovery Toolkit
Open Drug Discovery Toolkit (ODDT) is a modular and comprehensive toolkit written in Python for use in cheminformatics, molecular modeling. It does not take advantage of a well-known deep learning library and it requires only SK-learn.
ODDT includes many state-of-the-art methods, like machine learning scoring functions (RF-Score and NNScore). The goal is to create a tool that other people can customize for their computer-aided drug discovery. Apart from the tutorial linked in the repository, there is a reference paper published in the Journal of Cheminformatics. To manage and translate molecular data it requires installed the already mentioned RDkit or Openababel.
As interesting features, it includes 3 Molecular shape comparison methods (USR, USRCAT, and Electroshape)
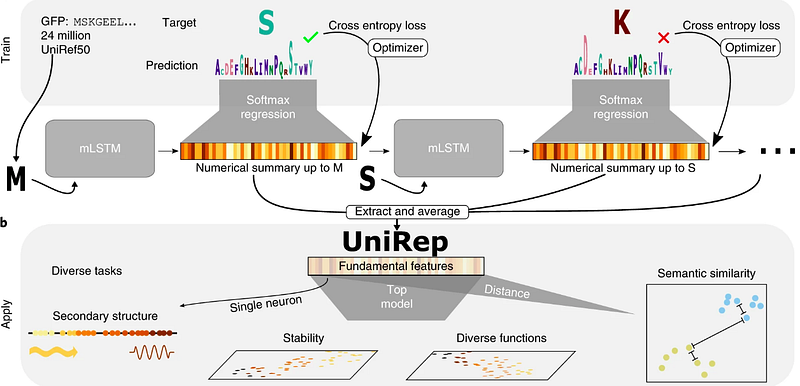
Unified rational protein engineering with sequence-based deep representation learning (UNIREP)
Unified rational protein engineering with sequence-based deep representation learning (Unirep) is a deep learning architecture which from unlabeled amino-acid sequences and phytochemistry can predict features of a protein that are semantically, evolutionarily, structurally, and biophysically grounded. It is an approach based on LSTM (mLSTM), a recurrent neural network architecture for sequence modeling that combines the long short-term memory (LSTM), which predicts the stability of natural and de novo designed proteins.
The code is freely available on GitHub, and the reference paper was published on Nature Methods 2019.
Conclusions
Until now, many pharmaceutical companies are still experiencing difficulties in drug design due to the high costs and time needed. Computational tools, especially those based on machine learning holds the promise to facilitate this process. A simple high-throughput screening library usually uses about 1 million compounds, where each compound typically costs 50–100 USD. Doing the same in-silico requires just a computer with a good graphic card, you can do the math.
Nevertheless, it is important to bear in mind that deep learning models are still something like a black box. A tremendous amount of work has been done to incorporate AI tools to expedite the drug discovery cycle, but further successful implementations of these tools will be necessary before the full potential of AI in drug discovery can be realized. It is highly appreciated that even commercial companies offer open-source tools and are open-minded to have communities expanding their tools.
CONNECT IF YOU WANT










{kind=link}